
一、基因组注释的概念及用处
基因组注释(Genomeannotation)是利用生物信息学方法和工具,对基因组所有基因的生物学功能进行高通量注释,是当前功能基因组学研究的一个热点。[1]即在一条DNA序列上,通过从头、同源、结构定义等多种方法,搜寻并定义基因组原件,得到其位置、序列、结构、功能等信息。[3]基因组注释的研究内容包括基因识别和基因功能注释两个方面。基因识别的核心是确定全基因组序列中所有基因的确切位置。[1]
解读遗传信息之二——基因组注释[15]
二、基因组注释相关背景知识
2.1基因组注释GFF>F文件[6][14]
GFF格式是Sanger研究所定义,是一种简单的、方便的对于DNA、RNA以及蛋白质序列的特征进行描述的一种数据格式,比如序列的哪里到哪里是基因。GFF格式已经成为序列注释的通用格式。
基因组注释文件是包含GFF,GTF两种主要格式,用于高通量测序中对已经map到参考基因组的reads做注释。这些文件是将各物种的每个染色体编号,并将其每个碱基位点编号,然后人们将已知的元件区间用起始位点和终止位点记录。
这样就可以知道reads是落在哪个基因,转录本上,准确的是落在了基因内,基因间,内含子,外显子上,亦或是在正链还是负链上。进一步将各个区域的reads统计counts,算出每个基因或元件的RPKM和TPM用于后续分析。
GFF>F文件由tab键隔开的9列组成,每一列代表不同的信息,下面是各列的说明:
GFF(general feature format):用于基因组注释。
-
seqid :通常格式染色体ID或是contig ID。
-
source:注释的来源,一般指明产生此gff3文件的软件或来源数据库。如果未知,
.代表空。 -
type: 一般使用gene,repeat_region,exon,CDS,或SO对应编号等。
-
start:起始位置,从1开始计数(需要注意:bed文件从0开始计数)。
-
end:终止位置。
-
score:得分,注释信息可能性说明,可以是序列相似性比对时的E-values值或者基因预测是的P-values值。
.代表空 -
strand:
+表示正链,-表示负链,.表示不需要指定正负链,?表示未知. -
phase :仅对编码蛋白质的CDS有效,本列指定下一个密码子开始的位置。可以是0、1或2,表示到达下一个密码子需要跳过碱基个数。
-
attributes:包含额外属性的列表,格式为
tag=value,不同属性之间以;相隔。-
ID–注释信息的编号,在一个GFF文件中必须唯一
-
Name–注释信息的名称,可以重复;
-
Alias–别名
-
Parent–指明feature所从属的上一级ID。用于将exons聚集成transcript,将transripts聚集成gene
-
Note–备注
-
Dbxref–数据库索引
-
GTF(gene transfer format):用于对基因的注释。
- seqname: 通常格式染色体ID或是contig ID。
- source:注释的来源。,一般指明产生此gff3文件的软件或来源数据库。如果未知,.代表空。
- start:起始位置,从1开始计数。
- end:终止位置。
- feature :表示基因结构。CDS,start_codon,stop_codon是一定要含有的类型。
- score :得分,注释信息可能性说明,可用
.代替空。 - strand:链的正向与负向,分别用
+和-表示。 - frame:密码子偏移,可以是0、1或2。
- attributes:必须要有以下两个值:
- gene_id value: 表示转录本在基因组上的基因座的唯一的ID。gene_id与value值用空格分开,如果值为空,则表示没有对应的基因。
- transcript_id value: 预测的转录本的唯一ID。transcript_id与value值用空格分开,空表示没有转录本。
2.2不同的基因注释版本[11]
- 主流的基因注释版本有三种:RefSeq/Ensemble/UCSC
- Refseq=NCBI;Ensemble=Gencode
- Ensemble注释更全面,Refseq适合那些不那么复杂的注释
Refseq是由美国NCBI搞出来的,而ENSEMBL则是由欧洲EMBL-EBI搞出来的,所以这俩不是一回事,甚至可以说差别有点大。
2.2.1Gencode
官方介绍:GENCODE项目的目标是基于生物学证据高精度地识别和分类人类和小鼠基因组中的所有基因特征,并发布这些注释以利于生物医学研究和基因组解释
Gencode的注释来源于两部分。分别是Ensembl-Havana团队生成的手动基因注释和Ensembl-genebuild的自动基因注释。当我们使用Ensemblgenomebrowser时,默认的基因注释就是Gencodeannotation。
这里值得一提的是,在gencode中标识HAVANA来源的,这表示它是人工注释的。但是这些注释也有可能是由于Havana手动注释和Ensembl自动注释合并的结果而如果标识的是ENSEMBL,则表明这条注释是由Ensemble自动注释得到的
实际上,GENCODE注释与Ensembl注释基本相同……此处划重点,是基本,但不是全部~那么有哪些不同呢?
-
X/Y染色体上PAR区域的注释:该区域的注释在Gencode中X染色体和Y染色体各注释一次,而在Ensembl文件中,只在X染色体进行了注释
PARregion(Pseudoautosomalregion):该区域是X和Y染色体的同源序列,因为这上面的任何基因可以和常染色体基因一样正常遗传而得名
-
Gencode的第九列,也就是attribute那一列,有一些其他额外的tags,这些tags是Ensebl所没有的
所以说gencode的基因组注释基本上和Ensemble是一样的。
2.2.2Gencode与Refseq
gencode的注释,我们最常用的是Comprehensive版本,这个版本有一个特点,那就是全。这个版本与Refseq相比,转录本注释有着更多更全的外显子,对基因组的覆盖范围更广,能够帮助我们发现更多的突变。
当然了,有Comprehensive版本,那一定有basic版本,就是下面这个了。这个版本与Refseq相比,相似性更高,没有什么所特有的features。

说了这么多,你可能要问了,不同来源的注释文件是否可以相互转换?那我只能很遗憾的告诉你:不能。虽然从整体上来看,这两个来源的注释信息大体上是相同的(如果只看某一些基因还是可以发现不同指出),但是这两个来源的注释都有不同的版本,而这些不同的版本很难一一对应。
不过,虽说注释信息不可以相互转换,但是RefseqID和ENSEMBL(Gencode)ID是可以相互转换的。我们可以使用在线工具http://www.ensembl.org/biomart/martview,也可以使用Biocundutor包调用API,比如biomaRT
2.2.3如何选择适合自己的注释文件?
首先我们需要知道,没有任何一个注释文件是完美的,总会有一些小的瑕疵。可能一些基因的注释不是很精确,甚至有可能全部都是错的……
如果我们想要做一些可重复度高的,或者对基因表达水平进行估计的时候,我们应该选择那些不那么复杂的基因注释,比如Refseq。当我们想要做一些探索性研究的时候,比如可变剪切之类的,我们可以选择比较复杂的基因注释,比如Ensembl

需要注意的是**.abinitio**后缀的GTF文件包括了Genscan和其他工具预测得到的注释信息,会全面一些。但是未必可靠!
Forthepredictedgeneset,anadditionalabinitioflagisaddedtothenamefile. 但是我自己选择的话,我倾向于使用Homo_sapiens.GRCh38.92.chr_patch_hapl_scaff.gtf.gz,因为在比对的时候fasta文件里面是带有scaffold的信息的。
需要注意,GENCODE上的GTF文件和ENSEMBL的GTF文件的第一列是不一样的,GENCODE的chr1在ENSEMBL上就是1。其余的地方是一样的(针对human&mouse而言)。
2.2.4问题
在比对的时候,我该选用哪一个类型的fasta文件呢?
RepeatMasking 在NGS处理过程中,事实上我们并不需要使用一个标记重复区域的基因组。标记重复的基因组会用N代替重复区域,而这就给后续的比对带来很大的问题,所以要避免使用**dna_rm-Repeatsmasked(convertsrepeatstotoN’s)的参考基因组。而dna_sm-Repeatssoft-masked(convertsrepeatnucleotidestolowercase)**虽然也标记出了参考基因组,但是以小写的形式存在的,故对比对没有影响。 这个问题问完了,dna_rm-Repeatsmasked出局,余下两者进入下一个问题。
PrimaryorToplevel?
简单的回答就是说请选择的primary版本,因为toplevel版本会包含haplotype信息,多余的信息会增加比对工具的工作,所以这里选择primary就可以完成你的工作。
多出来的chr_unkonw…..的信息是啥?
这是在构建基因组时已知存在于基因组内但是不知道位于哪条染色体上,随着注释工作的进展这些未知基因会越来越少。
三、基因组注释的五个研究方向
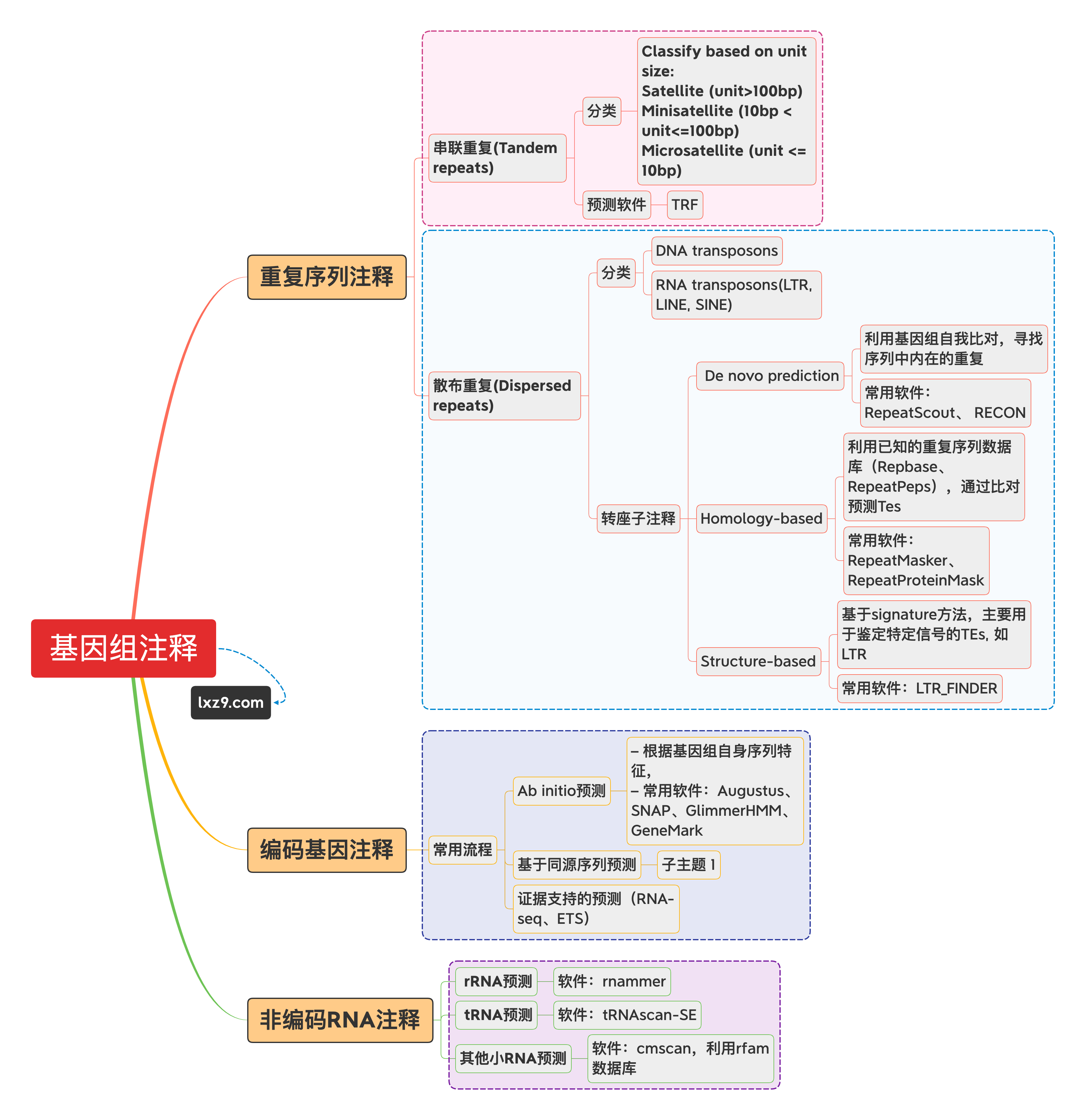
基因组注释主要包括四个研究方向:重复序列的识别、非编码RNA的预测、基因结构预测和功能基因注释。[1]此外还有假基因注释方面的工作。
3.1基因结构注释[2][8]
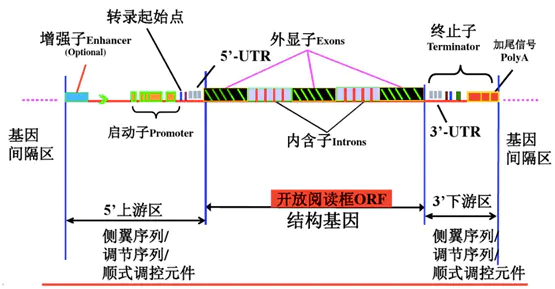
基因结构预测包括预测基因组中的基因位点、开放性阅读框架(ORF)、翻译起始位点和终止位点、内含子和外显子区域、启动子和终止子、可变剪切位点以及蛋白编码序列(CDS)等。需要指出,真核生物基因结构注释难度较大,主要因为真核生物中的启动子和终止子等信号位点较为复杂,且存在广泛的可变剪切现象,预测真核生物的基因结构常用隐马科夫模型。
基因结构注释采用从头测序,同源预测和基于RNA-Seq的证据支持预测相结合的方法。利用物种已发表的基因序列,蛋白序列,mRNA/ESTs序列集构建物种的基因结构模型;同时采用从头测序方法对初始预测模型进行自我训练,通过多轮训练和优化,获得从头预测的基因结构模型;利用RNA-Seq数据通过Tophat比对得到基因组的内含子结构模型及基因侧翼序列信息;最后对上述不同方法预测的结构模型进行整合和优化获得最终的基因结构模型。
注释之前首先得构建基因模型,有三种策略[9]:
- 从头注释(denovoprediction):通过已有的概率模型来预测基因结构,在预测剪切位点和UTR区准确性较低
- 同源预测(homology-basedprediction):有一些基因蛋白在相近物种间的保守型搞,所以可以使用已有的高质量近缘物种注释信息通过序列联配的方式确定外显子边界和剪切位点
- 基于转录组预测(transcriptome-basedprediction):通过物种的RNA-seq数据辅助注释,能够较为准确的确定剪切位点和外显子区域。
其中,从头预测主要应用软件有Augustus、Genscan、Glimmer等;同源预测代表软件包括Genewise(动物);而基于转录组数据预测则是由常见的Tophat+cufflinks软件完成。
3.1.1基因结构预测的研究背景和意义
通过基因结构预测,我们能够获得基因组详细的基因分布和结构信息,也将为功能注释和进化分析工作提供重要的原料。基因结构预测包括预测基因组中的基因位点、开放性阅读框架(ORF)、翻译起始位点和终止位点、内含子和外显子区域、启动子、可变剪切位点以及蛋白质编码序列等等。
3.1.2基因结构预测的发展现状
原核生物基因的各种信号位点(如启动子和终止子信号位点)特异性较强且容易识别,因此相应的基因预测方法已经基本成熟。Glimmer是应用最为广泛的原核生物基因结构预测软件,准确度高。而真核生物的基因预测工作的难度则大为增加。
- 首先,真核生物中的启动子和终止子等信号位点更为复杂,难以识别。
- 其次,真核生物中广泛存在可变剪切现象,使外显子和内含子的定位更为困难。因此,预测真核生物的基因结构需要运用更为复杂的算法,常用的有隐马尔科夫模型等。常用的软件有Genscan、SNAP、GeneMark、Twinscan等。
3.1.3基因结构预测的研究内容
基因结构预测主要通过序列比对结合从头预测方法进行。
- 序列比对方法采用blat和pasa等比对方法,将基因组序列与外部数据进行比对,以找到可能的基因位置信息。常用的数据包括物种自身或其近缘物种的蛋白质序列、EST序列、全长cDNA序列、unigene序列等等。这种方法对数据的依赖性很高,并且在选择数据的同时要充分考虑到物种之间的亲缘关系和进化距离。
- 基因从头预测方法则是通过搜索基因组中的重要信号位点进行的。常用的软件有Genscan、SNAP、Augustus、Glimmer、GlimmerHMM等等。同时采用多种方法进行基因预测将产生众多结果,因此最后需要对结果进行整合以得到基因的一致性序列。常用软件有Glean,EVM等。
3.1.4基因结构预测中拟解决的关键技术难点
目前,真核生物的基因结构预测方法仍有较大改进空间,主要面临以下的技术难点。
- 如何利用现有的数据和算法,更好地识别基因的可变性剪切位点。
- 随着测序工作的进展,许多目前研究较少的物种也将提上测序日程。大多基因结构的从头预测算法需要预先训练预测参数。现有资源和数据稀缺的物种将很难获得预测参数。
- 克服组装错误对基因结果预测的影响
3.1.5建立基因结构预测的评价系统
可变性剪切位点的预测较为困难。如何结合RNA-seq数据进行可变剪切预测将是重要的工作方向和难点。
3.1.6基因结构预测的研究方向
- 利用RNA-seq、EST等数据校正基因结构预测结果,识别可变剪切位点。
- 对于研究较少的物种,建议利用近缘物种的同源基因数据以训练基因结构预测软件。
- 利用同源基因组之间的共线性信息,辅助基因结构预测。
3.2基因功能注释[2][8]
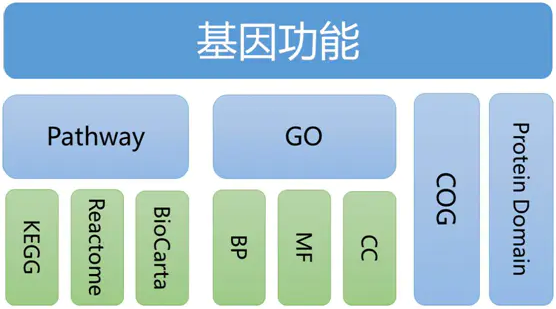
全基因组测序将产生大量数据,此前普遍采用比对方法对对预测出来的编码基因进行功能注释,通过与各种功能数据库(NR、Swiss-Prot、GO、KOG、KEGG)进行蛋白质比对,获取该基因的功能信息。其中GO和KEGG数据库分别在基因功能和代谢通路研究中占据重要地位。
3.2.1基因功能注释的研究背景和意义
获得基因结构信息后,我们希望能够进一步获得基因的功能信息。基因功能注释方向包括预测:
- 基因中的模序和结构域
- 蛋白质的功能
- 所在的生物学通路等
3.2.2基因功能注释的发展现状
全基因组测序将产生大量数据,而实验方法由于成本较高,不适用于全基因组测序的后续功能分析。为此,目前普遍采用比对方法对全基因组测序的基因功能进行注释。KEGG和GeneOntology是目前使用最为广泛的蛋白质功能数据库,分别对蛋白质的生物学通路和功能进行注释。Interpro通过整合多个记录蛋白质特征的数据库,根据蛋白质序列或结构中的特征对蛋白质进行分类。
3.2.3基因功能注释的研究内容
目前,我们利用四个常用的数据库进行基因功能注释。使用的数据库有Uniprot蛋白质序列数据库、KEGG生物学通路数据库、Interpro蛋白质家族数据库和GeneOntology基因功能注释数据库。
- 与Uniprot蛋白质序列数据库比对,获得序列的初步信息。
- 与KEGG数据库比对,预测蛋白质可能具有的生物学通路信息。
- 与Interpro数据库比对将获得蛋白质的保守性序列,模序和结构域等。
- 预测蛋白质的功能。Interpro进一步建立了与GeneOntology的交互系统:Interpro2GO。该系统记录了每个蛋白质家族与GeneOntology中的功能节点的对应关系,我们通过此系统便能预测蛋白质执行的生物学功能。
3.2.4基因功能注释中拟解决的关键技术难点
目前我们的功能注释工作是建立在比对的基础上,这将会带来两个比较大的问题。
- 首先,此方法严重依赖于外部数据,对某些研究较少的物种限制很大。
- 其次,序列相似并不表示实际生物学功能相似,考虑引入序列比对之外的方法,进一步完善基因功能注释工作。
3.2.5基因功能注释的研究方向
考虑引入序列比对之外的数据(如蛋白质互作网络、基因表达谱等),利用概率模型算法进行整合,完善基因功能注释工作。
3.3重复序列分析[2][8]
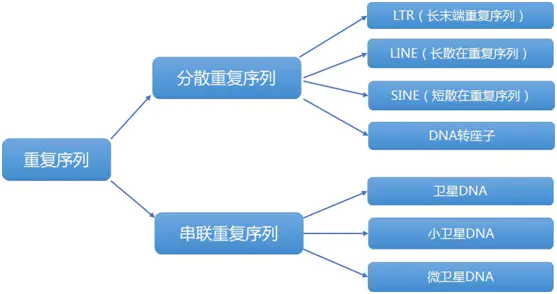
重复序列广泛存在于真核生物基因组中,这些重复序列或集中成簇,或分散在基因之间,根据分布把重复序列分为分散重复序列(Interpersedrepeat)和串联重复序列(Tendamrepeat)。重复序列的注释主要通过同源注释和从头注释两种方式进行预测。同源注释采用RepeatMasker通过与Repbase数据库进行比对寻找基因组中的重复区域,并对其进行分类;从头注释采用RepeatModler鉴定重复元件,最后通过整合获得全基因组的重复序列注释,从头注释能够发现未知的新的转座子元件。
3.3.1重复序列的研究背景和意义
重复序列可分为:
- 串联重复序列(Tendamrepeat),包括有微卫星序列,小卫星序列等等;
- 散在重复序列(Interpersedrepeat),散在重复序列又称转座子元件,包括以DNA-DNA方式转座的DNA转座子和反转录转座子(retrotransposon)。常见的反转录转座子类别有LTR,LINE和SINE等。
3.3.2重复序列识别的发展现状
目前,识别重复序列和转座子的方法为
- 序列比对
- 从头预测两类。
序列比对方法一般采用Repeatmasker软件,识别与已知重复序列相似的序列,并对其进行分类。常用Repbase重复序列数据库。
从头预测方法则是利用重复序列或转座子自身的序列或结构特征构建从头预测算法或软件对序列进行识别。从头预测方法的优点在于能够根据转座子元件自身的结构特征进行预测,不依赖于已有的转座子数据库,能够发现未知的转座子元件。常见的从头预测方法有Recon,Piler,Repeatscout,LTR-finder,ReAS等等。
3.3.3重复序列识别的研究内容
获得组装好的基因组序列后,我们首先预测基因组中的重复序列和转座子元件。
- 一方面,我们采用RepeatScout、LTR-finder、TendemRepeatFinder、Repeatmoderler、Piler等从头预测软件预测重复序列。为了获得从头预测方法得到的重复序列的类别信息,我们把这些序列与Repbase数据库比对,将能够归类的重复序列进行分类。
- 另一方面,我们利用Repeatmasker识别与已知重复序列相似的重复序列或蛋白质序列。通过构建Repbase数据库在DNA水平和蛋白质水平的重复序列,Repeatmasker能够分别识别在DNA水平和蛋白质水平重复的序列,提高了识别率。
3.3.4重复序列识别的关键技术难点
- 第二代测序技术测基因组,有成本低、速度快等优点。但是由于目前产生的读长(reads)较短。由于基因组序列采用kmer算法进行组装,高度相似的重复序列可能会被压缩到一起,影响对后续的重复序列识别。
- 某些高度重复的序列用现有的组装方法难以组装出来,成为未组装reads(unassembledreads)。有必要同时分析未组装reads以得到更为完整的重复序列分布图。之前,华大已开发了ReAS软件,专门用于识别未组装reads中的重复序列。但该软件目前只能处理传统测序技术(如sanger测序)生成的较长片段的reads,需要进一步改进方可用于分析第二代测序技术得到的reads。同时,未组装的短片段reads重复度更高,识别其重复区域具有较大难度。
3.3.5重复序列识别的研究方向
- 整合现有的重复序列预测方法,对组装好的基因组序列进行分析。
- 综合考虑并结合短序列组装策略,校正重复序列识别的结果。
- 开发识别未组装reads重复序列的算法和流程并构建一致性序列。
3.4非编码RNA注释[2][8]
非编码RNA,指不翻译成蛋白质的RNA,如tRNA,rRNA等。利用tRNAscan-SE对全基因组进行tRNA预测;利用RNAmmer预测全基因的核糖体RNA;利用Rfam数据库通过cmscan鉴定全基因组non-codingRNA(ncRNA)。
通过基因组注释获得的信息可进一步用于后续比较基因组分析,例如系统发育分析、基因家族分析、历史群体结构分析等,重复序列的注释则通常可用于全基因组加倍事件分析。但我们目前的大部分注释工作主要建立在与已有数据库的比对基础上,因此,对某些研究较少的物种限制很大。另一方面,序列相似并不表示实际生物学功能相似,这对于基因功能注释时会造成较大影响,仍需要进一步完善基因功能注释工作。
3.4.1非编码RNA预测的研究背景和意义
非编码RNA,指的是不被翻译成蛋白质的RNA,如tRNA,rRNA等,这些RNA不被翻译成蛋白质,但是具有重要的生物学功能。
- miRNA结合其靶向基因的mRNA序列结合,将mRNA降解或抑制其翻译成蛋白质,具有沉默基因的功能。
- tRNA(转运RNA)携带氨基酸进入核糖体,使之在mRNA指导下合成蛋白质。
- rRNA(核糖体RNA)与蛋白质结合形成核糖体,其功能是作为mRNA的支架,提供mRNA翻译成蛋白质的场所。
- snRNA(小核RNA)主要参与RNA前体的加工过程,是RNA剪切体的主要成分。
3.4.2非编码RNA预测的发展现状
由于ncRNA种类繁多,特征各异,缺少编码蛋白质的基因所具有的典型特征,现有的ncRNA预测软件一般专注于搜索单一种类的ncRNA,如tRNAScan-SE搜索tRNA、snoScan搜索带C/D盒的snoRNAs、SnoGps搜索带H/ACA盒的snoRNAs、mirScan搜索microRNA等等。Sanger实验室开发了Infernal软件,建立了1600多个RNA家族,并对每个家族建立了一致性二级结构和协方差模型,形成了Rfam数据库。采用Rfam数据库中的每个RNA的协方差模型,结合Infernal软件可以预测出已有RNA家族的新成员。Rfam/Infernal方法应用广泛,可以预测各种RNA家族成员,但是特异性较差。
我们建议:如果有更好的专门预测某一类非编码RNA的软件,那么采用该软件进行预测;否则,使用Rfam/Infernal流程。
3.4.3非编码RNA预测的研究内容
利用Rfam家族的协方差模型,我们采用Rfam自带的Infernal软件预测miRNA和snRNA序列。由于rRNA的保守性很强,为此我们用序列比对已知的rRNA序列,识别基因组中的rRNA序列。tRNAscan-SE工具中综合了多个识别和分析程序,通过分析启动子元件的保守序列模式、tRNA二级结构的分析、转录控制元件分析和除去绝大多数假阳性的筛选过程,据称能识别99%的真tRNA基因。
3.4.4非编码RNA预测中拟解决的关键技术难点
识别非编码RNA的假基因:基因组中很多序列由非编码RNA基因复制而来,与非编码RNA基因序列相似,但不具有非编码RNA的功能。目前我们采用的非编码RNA序列的预测方法都是基于序列比对和结构预测,不能够很好的去除这类非编码RNA的假基因。针对这个问题,我们考虑结合RNA表达信息如RNA-seq数据进行筛选。
3.4.5非编码RNA预测的研究方向
- 专门检测小片段RNA序列的方法现在已经得到广泛应用,利用小片段RNA序列数据进行非编码RNA的预测是我们的重要研究方向。
- 开发miRNA靶向基因预测流程:miRNA通过调控其靶向基因的mRNA稳定性或翻译来控制生命活动的进程。预测miRNA靶向基因能够给我们研究miRNA功能带来提示。由于miRNA在动物和植物中对靶向基因的调控机制差别较大,我们建议对动物和植物分别建立靶向基因预测流程,提高预测准确度。
3.5 假基因注释[12]
3.5.1 假基因注释的研究背景和意义
假基因是功能基因的缺陷拷贝,它源于蛋白质编码基因、与起源基因非常相似,但是不能编码蛋白质.假基因的形成,即基因正常活性的丧失是由对基因表达有阻断作用的突变导致的.这些变化主要包括消除起始转录的信号,阻止外显子/内含子连接点的剪接或过早地终止翻译等.研究假基因首先是因为他可以为进化研究提供参考,另外就是“死去的基因”被证明也是有功能的。
3.5.2 假基因注释的发展现状
在过去几年时间里,有关假基因的研究取得了很大的进展,主要集中在假基因的来源,假基因的识别,进化研究,分布情况和功能上。对于基因组注释我们只需关注如何进行正确的预测和识别。
3.5.3 假基因注释的研究内容
正如上文所说,假基因的鉴别是假基因相关研究的基础,而且它对基因注释的精确度也有很大影响.尤其是基因与重复假基因之间的区分是很有挑战性的问题.假基因的识别工作主要是由耶鲁大学Gerstein实验室研究小组完成的.他们从果蝇、线虫、小鼠、人等很多物种基因组中系统地搜索识别假基因,并创建了专门的假基因数据库(http://www.pseudogene.org),可供研究人员免费下载使用。表1中列举了目前国际上几个通用的假基因数据库[21-23]和假基因识别程序.
3.5.4 假基因产生的两种途径
产生假基因的渠道主要有以下两种[2-3]:一是基因组DNA重复或染色体不均等交换过程中基因编码区或调控区发生突变(如碱基置换、插入或缺失),导致复制后的基因丧失正常功能而成为假基因,这种假基因称为重复假基因(duplicated pseudogene);二是mRNA转录本反转录成cDNA后重新整合到基因组,由于插入位点不合适或序列发生突变而失去正常功能,这样形成的假基因称为加工假基因或返座假基因(processed pseudogene or retro pseudogene)
四、基因组注释的操作流程[3][8]
如何对基因组序列进行注释[9]
基因组结构注释[10]
如何对基因组序列进行注释[13]
所有的基因组注释都会依赖流程化的计算过程,而对于参考基因组来说,往往还需要手工来校正。手工校正被看作是一项金标准,这也是一个高标准注释项目的核心程序之一。就像我们前面所说的,流程化的计算过程需要三个方面的资源,即转录组,相近物种的基因注释集,以及从头预测。而从头预测在高等真核生物里已经用的非常少了。一些超大型的基因组项目需要自己注释一份基因集以期能够更好的回答科学问题。出于一些实际的原因,有些研究者只是单纯的利用二代测序拼装完成的转录组序列来注释基因组,因为这种方法错误率太高,所以我们并不认同这种做法。[6]
4.0基因组注释前期准备
物种拉丁名,例如:Orazysativa,基因id:Osa000001 同源物种:一般选5个左右物种,需要有注释的基因/蛋白序列,保证高组装和注释质量 转库组数据:RNAseq和lsoseq注释(用于结构注释中的转录辅助注释)(建议自测同样本的数据)
4.1重复注释
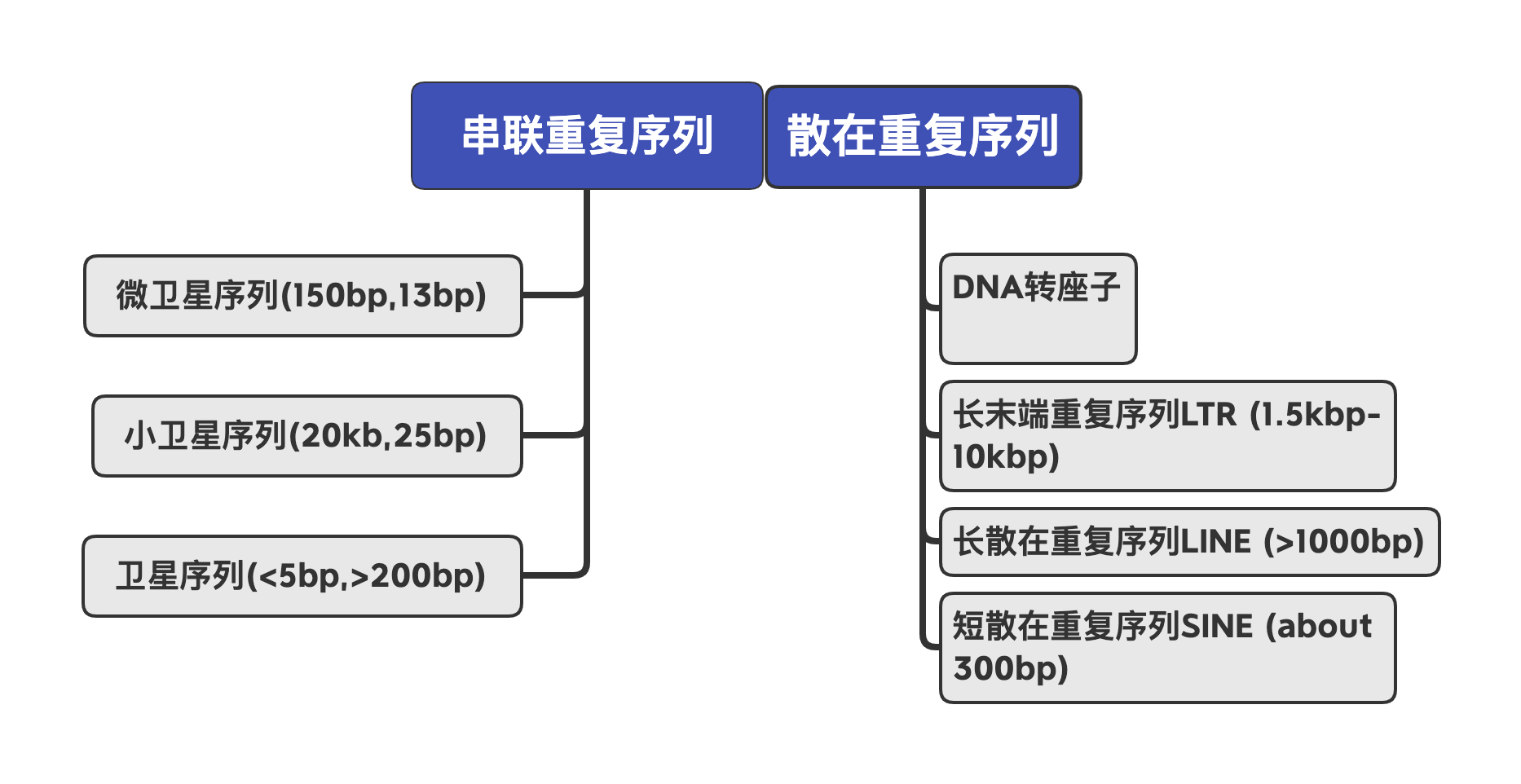
重复序列广泛存在于真核生物基因组中,这些重复序列或集中成簇,或分散在基因之间。根据分布把重复序列分为散在重复序列和串联重复序列。 重复序列根据序列特征分为2类:串联重复(Tandemrepeats)和散布重复(Dispersedrepeats)
-
RepeatMasker:基于Repbase(dna)/自建elibrary查询重复序列
``` BASH RepeatMasker-nolow-no_is-norna-parallel2-libRepeatMasker.libgenome.fa #-nohow:屏蔽低复杂简单重复;-no_is:跳过细菌插入元件检查;-norna:不掩盖小RNA(伪)基因; #-parallel并行使用的处理器数,可提升分析速度 ``` -
RepeatProteinMask:基于Repbase(pep)查询重复序列
``` BASH RepeatProteinMask-noLowSimple-pvalue0.0001genome.fa #noLowSimple:关闭低复杂度和简单重复的屏蔽/注释;-pvalue:接受匹配的阈值 #注意点:genome.fa的D不能长于18个字符 ``` -
TRF:元件的结构特征等来识别重复序列
``` BASH trfgenome.fa2778010502000-d-h ``` -
LTR-FINDER:基于重复序列特征
``` BASH Itr_finder-W2-C-stRNAs.fagenome.fa #-w2输出格式,2-table;-C:检测中心粒,删除高重复区域 ``` -
repeatmodeler:基于自身序列比对
``` BASH BuildDatabase-namemydbgenome.fa RepeatModeler-databasemydb-pa6>run.out #-name:创建database的名称; #-pa:共享内存处理器的数量程序,可提升分析速度 ```
每个软件都有很多参数,可-help/-h自行查看,参数的选择最好是参考已发表的文献
4.2结构注释
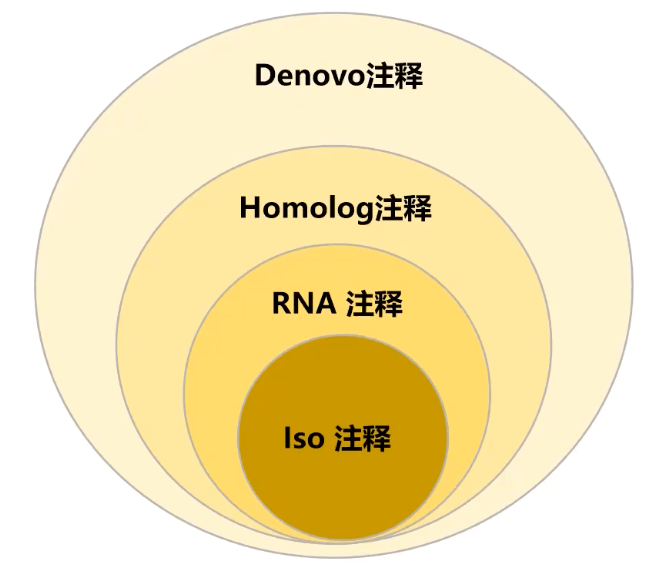
结构注释:注释可以产生具有生物学功能的蛋白的基因。一般包括启动子,转录起始,5’UTR,起始密码子,外显子,内含子,终止密码子,3’UTR,poly-A等结构。
4.2.1Denovo预测(屏蔽重复序列)
-
Augustus(真核)
BASH augustus--species=XXX--AUGUSTUSCONFIGPATH=config--uniqueGeneld=true--nolnFrameStop=true--gff3=on--strand=bothgenome.mask.fa>genome.mask.fa.out #--uniqueGeneld=true:gene:命名aseqname.gn; #--nolnFrameStop=true:不带有终止密码子的转录本; #--gff3=on:输出格式gff3 -
GlimmerHMM(真核,预测的基因数目较多长度较短,一般用于植物)
BASH glimmerhmm.genome.mask.fa-dXXX-f-ggenome.mask.fa.gff #-d库de路径; #-f:不要partialgenepredictions; #-g输出格式gff -
Genscan(真核,其预测的内含子较大,一般用于动物)
BASH genscanHumanlso.smatgenome.mask.fa>genome.mask.fa.genscan #Humanlsc.smat:参数文件,软件自带 -
其他软件
SNAP.GenelDGenemarkS denovo的软件很多,两个软件就可以了,太多软件会增加较多的假阳性,一般在Augustus,GlimmerHMM,Genscan中选择即可
4.2.2Homolog注释
利用近缘物种已知基因进行序列比对,找到同源序列。然后在同源序列的基础上,根据基因信号如剪切信号、基因起始和终止密码子对基因结构进行预测。 相对于从头预测的“大海捞针”,同源预测相当于先用一块磁铁在基因组大海中缩小了可能区域,然后从可能区域中鉴定基因结构。
利用TBlastn将同源物种的蛋白比对回基因组,得到候选区域。 利用EXonerate/Genewise进行精确的蛋白-核酸比对,以得到剪接位点。 Exonerate解决了GeneWisez存在的很多问题,并且速度快了1000倍,默认选择EXonerate分析
4.2.3RNA-seq辅助注释
tophat比对————>cufflink转录本————>TransDecoder
- 将RNAseq数据进行tophat比对;
- 比对后的结果文件利用cufflink构建转录本
- 使用TransDecoder在构建的转录本上预测OpenReadingFrame(ORF)。
4.2.4Isoseq辅助注释
CD-HIT————>gmap比对————>TransDecoder
- 将物种的三代全长转录本用CD-HIT进行去冗余;
- 将去冗余后的序列使用gmap比对回基因组得到转录本位置;
- 使用TransDecoder在构建的转录本上预测OpenReadingFrame(ORF).
基因结构预测方法可信度排序
4.2.5MAKERE整合
在基因组注释上,MAKER算是一个很强大的分析流程,主要是进行Denovo注释,Homolog注释,转录辅助注释三者的整合,保证最终注释基因集的可靠性
BASH
makermaker_exe.ctlmaker_opts.ctlmaker_bopts.ctl
#makerexe.ct:执行程序的路径
#maker_boots.ctl:BLAST7和Exonerate的过滤参数
#makeropts.ctl:其他信息,例如输入基因组文件,主要调整输入文件等(genome=;est=;protein=;pred_gff=;)
4.3nCRNA注释
- rRNA(核糖体RNA) 与蛋白质结合形成核糖体,其功能是作为mn的支架,提供mRNA翻译成蛋白质的场所。
- tRNA(转运RNA) ·携带氨基酸进入核糖体,使之在mRNA指导下合成蛋白质。
- miRNA(miRNA) ·将mRNA降解或抑制其翻译,具有沉默基因的功能。
- SnRNA(小核RNA) ·主要参与RNA前体的加工过程,是RNA剪切体的主要成分。
4.3.1miRNA与snRNA注释
- 采用Rfam和INFERNAL进行二级结构检测。
- ftp://ftp.sanger.ac.uk/pub/databases/Rfam
- blastn+cmsearch(INFERNAL程序)
4.3.2rRNA注释
- 由于rRNA的结构保守程度非常高,因此采用与已有的全长rRNA进行blastn比对而获得。
- blastn
4.3.3tRNA注释
- 结构特点:三叶草型二级结构。
- 预测方法:针对二级结构进行检测。使用tRNAscan-SE
4.4功能注释
功能注释:基因功能的注释依赖于上一步的基因结构预测,根据预测结果从基因组上提取翻译后的蛋白序列和主流的数据库进行blastp比对,完成功能注释。
常用数据库一共有以下几种:NR,KEGG,Uniprot(Swiss-Prot,TrEMBL),InterPro,Go
-
KEGG
-
生物学通路数据库(Gene,Pathway,Ligand).
-
http://www.genome.jp/kegg/
-
blastp
-
-
SWISS-PROT和TrEMBL
-
UniProt(UniversalProteinResource)蛋白质序列数据库PIR、SWISS-PROT和TrEMBL统一起来,建立了一个蛋白质数据库。
-
http://www.uniprot.org/
-
blastp
-
-
Interpro
-
蛋白家族(proteinfamilies)、功能保守区域(domains)和功能位点(funtionalsites)的数据库.
-
http://www.ebi.ac.uk/interpro/
-
InterProScan
-
-
GO
-
基因功能注释数据库(GeneOntology)
-
三个层面CellularComponent、BiologicalProcess、MolecularFunction.
-
http://www.geneontology.org/
-
InterProScan
-
4.5基因组评估
-
BUSCO评估
BUSCO是一款使用python语言编写的对转录组和基因组组装质量进行评估的软件。在相近的物种之间总有一些保守的序列,而BUSCO就是使用这些保守序列与组装的结果进行比对,鉴定组装的结果是否包含这些序列,包含单条、多条还是部分或者不包含等等情况来给出结果。 BUSCO软件根据OrthoDB数据库,构建了几个大的进化分支的单拷贝基因集。将其与该基因集进行比较,根据比对上的比例、完整性,来评价准确性和完整性。
五、基因组注释的操作细节
5.1基因组整理【组装之后,注释之前】
5.1.1基因组碱基整理
基因组碱基-u修改成大写字母形式,同时建议使用-w0把序列输出限定为一行序列。
seqkitseq-u-w0genome.old.fa>genome.new.fa
5.1.2基因组碱基小写带来的问题:
-
edta运行TIR预测时,报错并中断运行。
EDTA/bin/TIR-Learner2.5/Module2/RunGRF.py",line79,in<module> if(len(str(records[0].seq))>int(length)+500): IndexError:listindexoutofrange cp:cannotstat'TIR-Learner/*-p':Nosuchfileordirectory -
geta生成基因注释结果减少。 前期运行不受影响,会在geta生成的/out.tmp/6.combineGeneModels/中合并前面步骤预测结果时把许多基因预测结果识别为partial而筛除掉本为完整的基因,可看到/out.tmp/6.combineGeneModels/genome.gff3的gene数量是正常的,而out.tmp/6.combineGeneModels/genome.completed.gff3明显降低,/out.tmp/6.combineGeneModels/genome.partial.gff3明显升高。out.tmp/6.combineGeneModels/genome.filter.gff3是最后的基因注释结果,即out.gff3也相应减少。
-
maker的某些运行步骤也会出错。
5.1.3基因组序列ID修改
(一)排序【optional】–根据需要选择
按长度对基因组contigs进行从长到短的排序
seqkitsort-l-r-w0genome.old.fa>genome.sort.fa
(二)重命名【推荐】
最好重命名成长度一致的数字,避免后期分析遇到contig1和contig11同时被匹配的问题。
-
solutionA【速度最快,推荐】 序列IDs按顺序命名为1-n的数字,–nr-width可以设置数字的长度,长度为5时第一条序列名称为00001。
seqkitreplace-p'.*'-r{nr}--nr-width5genome.sort.fa -
solutionB 把序列id重命名成1-n的数字
awk'BEGIN{i=0;FS=",";OFS=","}{if(/>/){gsub($1,">"++i,$1);print$0}else{print$0}}'genome.sort.fa>genome.fa基因组的序列id改为scaf001的形式
sed-i"s/>/>scaf/g"genome.fa -
solutionC 把基因组序列id的数字1-100改成0001-0100这种数字位数相同的格式。这个没有solutionA快,只推荐在不想做大规模更改的情况下使用。
用下面脚本实现:
``` #!/bin/bash j=1 foriin`seq-w10100`#把相同数字位数的0001-0100依次赋值给变量i do sed-i-E-e"s/scaffold$j$/scaffold_$i/g"-e"s/scaffold$j([0-9])/scaffold_$i\1/g"species.fa#两次替换,第一次替换scaffold$j为行尾的字符串(比如在基因组序列文件中),第二次替换scaffold$j不为行尾的字符串,[0-9]代表不为数字的任意一个字符,\1代表替换前括号([0-9])中的内容。 sed-i-E-e"s/scaffold$j$/scaffold_$i/g"-e"s/scaffold$j([0-9])/scaffold_$i\1/g"species.gff#同上,替换其他文件,比如gff文件。 j=$(($j+1))#把1-100依次赋值给变量j done ```
5.2基因组注释整理【注释结果整理】
5.2.1合并多个注释结果
基因组的基因结构注释,如果使用了多款软件进行,想要合并多套注释结果,并让注释的基因根据染色体和位置信息排序,可参考这个办法。
合并两个基因注释文件(若是多个就依次合并)
- 输出A.gff中符合要求的行,即A.gff中与B.gff中位置有overlap的行。
bedtoolsintersect-aA.gff-bB.gff-wa>A.dup.gff - 输出只在A.gff存在的行,即把A.dup.gff从A.gff中删除,需要两个文件都已排序,若未排序先sort排序再处理。
comm-1A.gffA.dup.gff>A.filter.gff - 合并B.gff和A.gff中与B.gff不重复的部分。
catB.gffA.filter.gff>sample.gff
5.2.2重命名注释结果【可选】
根据需要选择【非必要的】,根据染色体位置顺序对注释的基因结果进行排序和重命名整理。 除了基因注释外,不同分析源都可能发现新的基因,增加注释的基因数量,从而打乱顺序。
(一)排序
- 把sample.gff3中gene替换成1gene,mRNA替换成2mRNA,exon替换成3exon,CDS替换成4CDS;目的是确保排序后单个基因内部的顺序是1-2-3-4。
sed-i-e"s/gene/1gene/g"-e"s/mRNA/2mRNA/g"-e"s/exon/3exon/g"-e"s/CDS/4CDS/g"sample.gff3 - 根据染色体位置排序。按照第一列第四个字符,第四列数值,第五列数值逆序依次排序。
catsample.gff3|sort-k1.4n-k4n-k5nr>sample.sort.gff3 - 获得的sample.sort.gff3文件再1gene替换成gene,234类似替换。
sed-i-e"s/1gene/gene/g"-e"s/2mRNA/mRNA/g"-e"s/3exon/exon/g"-e"s/4CDS/CDS/g"sample.sort.gff3
(二)重命名
- 复制一份注释文件用于修改和替换
cpsample.sort.gff3sample.new.gff3 - 获取旧ID的list。获取sample.sort.gff3第九列中的ID值。
catsample.new.gff3|awk'$3=="gene"{print$9}'|sed-e"s/;.*//g"-e"s/ID=//g">old.name - 获取新ID的list。
foriin$(seq-w1catold.name|wc-l);doechoscaf$i;done>new.name - 合并旧的和新的ID。
paste-d"/"old.namenew.name>old2new.name - 生成替换脚本old2new.sh。
sed-e"s//sed-i\"s\//g"-e"s/$/\/gsample.new.gff3/g\""old2new.name>old2new.sh - 运行替换脚本,会直接替换sample.new.gff3文件的旧ID为新ID,可能会运行较长时间。
shold2new.sh
ps:这部分代码可实现,但是是非常冗余的代码量(水平有限),且未必适用所有注释文件,谨慎参考。
5.2.3从注释文件提取序列
用gffread根据注释文件从基因组提取基因序列:
- 从基因组genome.fa和注释文件sample.gff3获取cds序列
gffread-xsample.cds.fa-ggenome.fasample.gff3 - 从基因组genome.fa和注释文件sample.gff3获取exon序列
gffread-wsample.exon.fa-ggenome.fasample.gff3 - 从基因组genome.fa和注释文件sample.gff3获取protein序列
gffread-ysample.protein.fa-ggenome.fasample.gff3
5.2.4从注释文件提取intron信息
六、基因组注释的困难与挑战
6.1基因组注释的难点[7]
-
区分基因和假基因
-
可变剪切转录本是否编码并产生真正的蛋白
-
一些位于编码区的转录本并不能产生蛋白,那么它的使命是什么?
-
注释出的蛋白序列仍然需要蛋白水平上的证据
-
非编码RNA的注释。这一块的工作千万不要被组学水平上的一些鉴定所迷糊眼界。
-
基因之外。也即目前认为的基因间隔区,这些区域的生物学功能仍是研究的盲点。
-
基因组的可用性。获得的高质量基因集也需要建立一个方便的访问和使用接口。
6.2基因组注释的局限性[8]
随着测序技术的成熟以及测序成本的降低,对产生的越来越多的序列进行注释也显得尤为重要。微生物的基因组注释往往包括自动注释,以及之后的手动注释的矫正。大多说的注释用的是同源性方法,通过与相近的已知的参考基因组比对,比对上,则认为有相近的功能。自动注释容易产生低质量的注释结果和错误,而手动注释则是为了去掉这些错误。但现在测序产生的大量数据,如果再用手动注释则显得不靠谱了。
高质量的注释不光仅仅是用一个软件预测一下ORF,然后获取跟他比对结果最好的已知的参考基因的信息。还应该包括ribosomal-bindingsites(RBSs),terminationsitesandconservedmotifs/domains.上面的这些注释不仅是为了更全面的注释一个基因,还可以更正之前注释产生的错误。例如,通过RBSs和terminationsites可以更群出知道基因真实的位置,这些软件有。
如果仅仅依靠序列相似性来找注释有其局限性。我们获得一个新的序列,本来是想找它与其相近的序列的不同,却用相似性来注释它。而且如果参考基因组中没有相应的基因,也就可能漏掉我们感兴趣的区域。
随着数据量增加,如果仅凭注释出来的基因组的信息现在是很难上传到公共数据库中。而现在二代测序中的RNA-seq则是结合实验结果,这样能够给相应蛋白的角色和功能有个更好的注释。这样的注释往往更可信,因为他不仅仅是根据序列相似性,还有实验的证明。目前,基因组的注释信息应该包含注释信息的的来源以及可靠性的依据,但是这一般被自动注释给省略了。包含证据信息可以给我们这注释信息的可靠性。
如果注释较真起来会有很多的问题。现在用来作为参考序列的数据也许是依赖更早的数据而得出的信息,错误的注释信息和错误也许能保留在新的基因组中,然后错误的信息最终带入到二代数据库中,例如UniProt,KEGGHE区域特异性数据库PATRIC.公共序列数据库认识到了控制错误复制和提供可靠的软件来检查用户上传的数据的需要。找到自动注释常见的一些错误,然后找方法来更正他们。
6.2.1注释的不一致性
同一个基因组用不同的工具注释出来的信息有时候也不同,常见的问题是分开/融合问题.
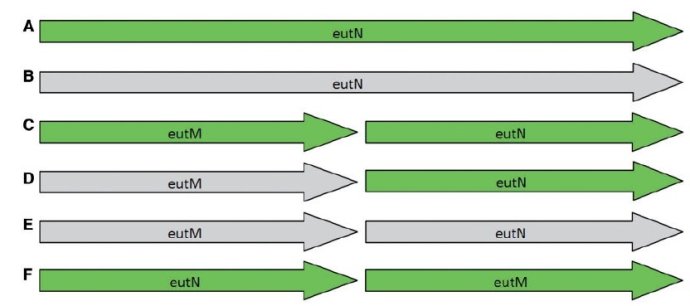
Thesixdifferentmodelspresentacross17RefSeqentriesforSalmonellaspeciesfortheeutM/eutNlocus.Greenindicatesnormalgene/CDSfeatures,grayindicatesgenefeaturesannotatedaspseudogenes.(A)Asingleintactgeneof690bp;(B)asinglepseudogeneof690bp;(C)twoshortintactgenes300bpinlength;(D)onepseudogeneandoneintactgene,each300bpinlength;(E)twopseudogenes,each300bpinlength;and(F)twointactgeneswiththeorderreversed.
说白了上图就是同一个基因在用不同的注释工具注释出来的结果不同,颜色浅代表是假设的注释结果。同名但是序列不一样。
直接预测区域(domains),而不是基因,用PfamAlyzer这个工具可能有助于找到区域中分开的基因。在上图的那个例子中,预测区域在所有的情况中会鉴别两个真实的区域,但是这个区域是来自一个还是来自两个基因则是没有办法弄清楚的。最正确的注释是这个区域是把基因组跟这6个不同模型都比对一下,但是自动注释操作起来还是会有很多问题,如果结合实验数据(RNA-Seq)就更好了。
还有一种情况,序列同源但是不同名,如下图
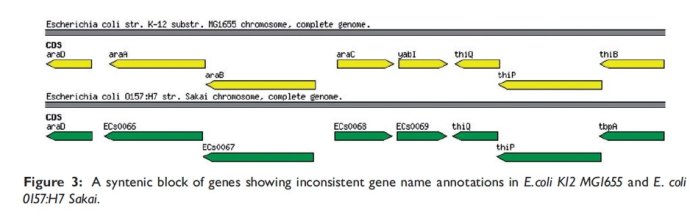
所以说嘛,参考序列选择的不一样,如果参考序列里面的注释信息不一致,那么注释出来的信息肯定也不一致,所以在选择相应的基因组来作为参考时,必须选择正确的包含最新信息的序列,同时未知的这个基因组不仅仅是跟一个参考基因组比较,跟其他的比较也是一样的。
6.2.2拼写错误
例如在UniProt中,有128个蛋白包含“syntase”,正确的应该是‘synthase‘.如果拿正确的拼写来搜索的话,会漏掉很多的信息。手动来改掉拼写错误是一个巨大的工作量,能否利用一些能够提示错误的计算机语言将会非常有帮助。
同一个基因名,不同产物名字
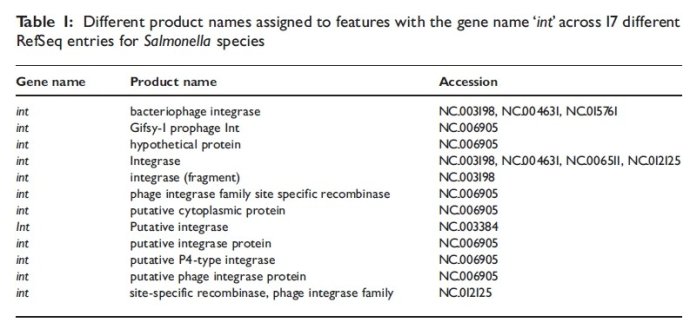
6.2.3假设蛋白
假设蛋白是那些被软件预测出来,但在数据库中找不到已知功能的同源基因,也找不到同源的已知功能域。他们可能是真实的未知功能的基因,也许是由于预测产生的错误。许多细菌未知功能的基因根据他们在E.coliK-12的相对位置来命名为y-gene。通常一些仅仅跟其他假设特征同源的特征,这些特征也灭有包含任何区域,现有的区域预测软件无法预测出来,是否注释这些特征也成为了一个思考的问题。正是基于这样,一些‘hypotheticalproteins‘被注释出来,发表和传播到新的数据库中,最后呈现在用户注释新的基因组的结果中。每个注释出来的特征的打分呈现出来也是非常有必要的,这样用户可以根据得分对可信度有一个初步的判断。
对是否把这些蛋白放在注释信息里面有争议,如果他们的确是因为基因预测产生的错误,他们就应该被删除,否则他们会一直保存在第二代或第三代数据库中直到该蛋白被证实有功能。用Pfam和InterPro来寻找保守的区域或模型能让我们知道这个假设的蛋白是否有功能,但是这也有缺陷。事实上,蛋白有区域得分高并不是一定就说明有功能。Pfam, for example, contains over 3000 ‘domains of unknown function’, or DUFs, representing over 20% of known families 。
仅凭计算方法去得出一个基因组的区域是否有功能没有实际的意义,未知功能的保守特征也应该保留,这些特征可能会含有我们感兴趣的信息;但是他们应该跟我们有足够证明的其他的特征分开比较以让人知道他们的确信度是有不同的。
6.2.4直系同源和旁系同源
同源意味着基因有一段共同的区段,直系同源来自基因的分化,旁系同源来自基因的复制。区分直系和旁系同源不仅对于进化关系至关重要,对功能关系也很重要,直系同源一般都保留相似的功能,而旁系同源随着时间的推移往往是具有不同的功能。
参考资料
[2]:基因功能 | 注释方法
[3]:如何进行 | 基因组注释
[4]:基因组组装和注释 | 结果的整理
[7]:再说 | 基因组注释
[8]:【1.1】 | 基因组注释
[9]:如何对基因组序列 | 进行注释
[10]:基因组 | 结构注释
[11]:基因 | 注释
[12]:干货: | 基因组注释详解
[13]:如何对基因组序列 | 进行注释
[14]:基因组注释文件(GFF,GTF) | 下载的四种方法
[15]:解读遗传信息之二—— | 基因组注释




暂无评论内容