
一、生信常用数据库
最常用 Uniport 数据库,第二优先级是 GeneBank/NCBI 数据库,最次选择 RNA sequence 序列信息进行氨基酸转换。[5]
1.1 Uniport
Uniport数据库中的Swiss model数据库是人工检查过的,uniprot数据库中的subcellular location提供了蛋白质亚细胞定位的信息,对于有信号肽的蛋白质定位于线粒体,没有信号肽的留在细胞核。Pathology & biotech提供蛋白质突变或缺失导致的疾病及表型信息;PTM/Processing提供蛋白质翻译后修饰(post-translational modification, PTM)或翻译后加工的相关信息;family & domains提供蛋白质家族及结构域信息。[1]
上面有各种物种(包括植物)的蛋白详细信息,包括序列,结构,功能介绍,详细的annotation和分类,甚至列了很多相关publication参考文献(当然很多时候不全,需要另外查询,但可以参考,有一些列出来的文献自己找还未必能找到),最后还有一个protein interaction network,这个很强大,它是通过text mining的方式查找(有些基于实验结果)和预测可能有相互作用的蛋白,虽然不一定实用,但是可以给出很多hints, 而且可以挖出一些很偏的文献。[2]
这是一个关于蛋白质序列和功能信息的高质量且易于访问的数据库,整合了Swiss-Prot、 TrEMBL 和 PIR-PSD 。许多蛋白序列都来自于基因组测序项目,而且它还包含了来自研究文献中蛋白质的生物学功能的大量信息。[3]
1.2 NCBI
网址:https://www.ncbi.nlm.nih.gov/
不用说了,有很多功能,blast找蛋白或同源蛋白,蛋白序列,保守domain。查看保守domain是我个人比较常用的功能。其他还的很多功能可以多了解一下,比如蛋白上与疾病相关的variation等,在dbVar子目录下。[2]
NCBI是一种比较初级序列信息的算法。BLAST搜索允许你将感兴趣的序列与数据库的同源性序列进行比较。它也可以用来对比不同类型的序列,如氨基酸或核苷酸。[3]
1.3 Ensembl
ensembl,前面已经说过了,信息非常丰富,序列,假基因,snp,utr,要啥有啥。里面工具很多,我们平常最常用的是http://web.expasy.org/protparam可以估计蛋白的pi和疏水性,做纯化用。[2]
Ensembl是一项生物信息学研究计划,旨在开发一种能针对参考基因组(reference genome)自动产生关于基因比对和其他数据的图形视图。Ensembl的一个特点就是你可实时打开或关闭某些功能或者依据你的兴趣进行信息追踪;同样,它在获取基因一些基本信息如转录长度、编码外显子数甚至有关GO的详细细节上,都是非常有用的。[3]
1.4 IPA
网址:https://digitalinsights.qiagen.com
IPA(Ingenuity Pathway Anaylsis)是一款基于云计算的一体化应用软件,可以分析、整合、理解来源于基因表达、microRNA、SNP微阵列、代谢组学、蛋白组学、RNA-Seq实验以及能生成基因、化合物表达的各类小规模实验数据。IPA数据库还包含各类基因、蛋白、化学分子、药物的靶标信息,并且可以构建实验系统下的相互作用模型。这个软件虽然不是免费的,但是却有一个免费试用期。[11]
1.5 Phosphosite
网址:https://www.phosphosite.org/homeAction.action
PhosphoSitePlus数据库是一个由 CST 和 NIH 联合开发的免费资源数据库,总结归纳了海量通过科学研究发现的蛋白修饰位点,包括磷酸化、甲基化、乙酰化、泛素化等,并且包括一些 CST 公司发现但未发表的蛋白修饰位点。[11]
该数据库是动态的、开放的、高度互动并持续更新的。它有助于研究 PTMs 在正常和病理细胞/组织中的作用,同时它也是发现新的疾病标志物和药物靶点的有力工具。[11]
- 在 PhosphoSitePlus数据库我们能够查询:
- 一个蛋白质存在哪些翻译后修饰位点
- 与特定蛋白修饰相关的疾病、细胞系、组织类型
- 蛋白的亚细胞定位、修饰蛋白序列、基因序列、结构域等
- 在蛋白的空间水平上显示蛋白修饰位点的位置
- 及时更新的激酶底物和相关序列
1.6 The Human Protein Atlas
网址:https://www.proteinatlas.org/
内含近30000种人类蛋白质的组织和细胞分布信息,并提供免费查询。瑞典Knut&Alice Wallenberg基金会利用免疫组化技术,检查每一种蛋白质在人类48种正常组织,20种肿瘤组织,47个细胞系和12种血液细胞内的分布和表达,其结果用至少576张免疫组化染色图表示,并经专业人员校对和标引,保证染色结果具有充分的代表性。
二、生信常用方法
2.1 一些数据可视化方法
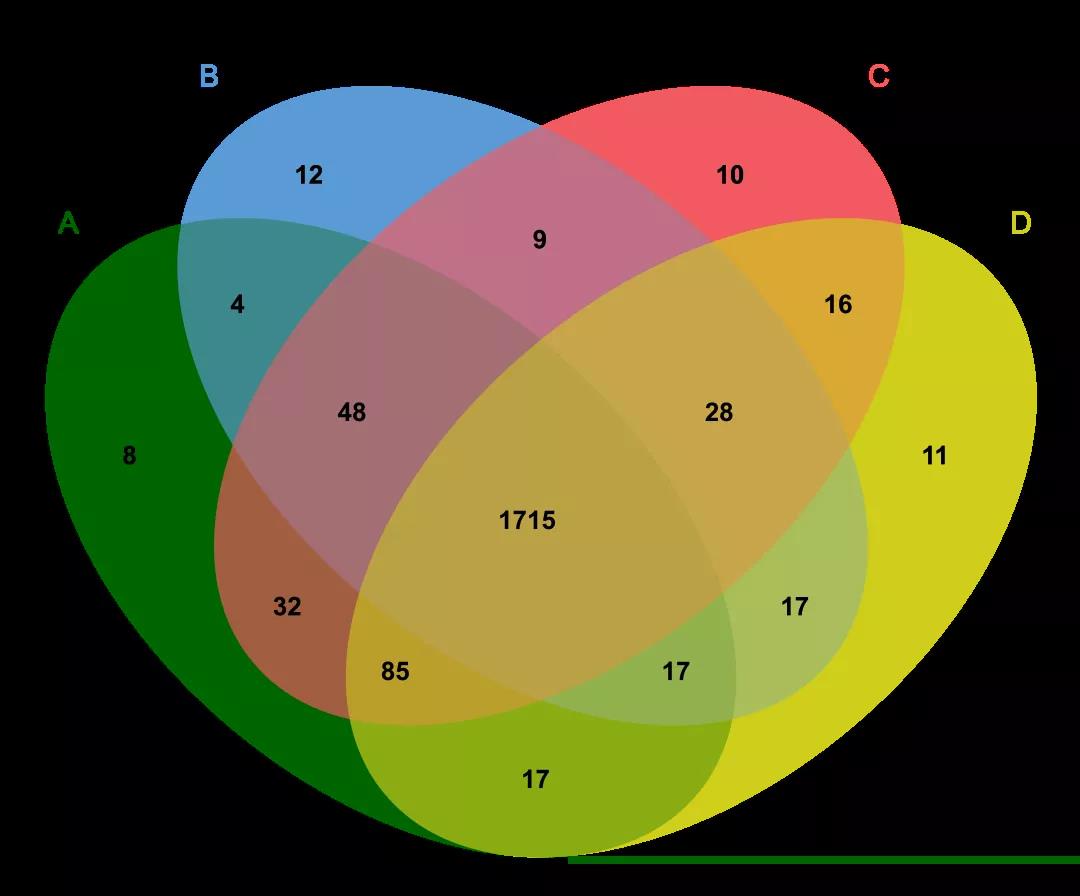
2.2.1 韦恩图[9]
不同处理组之间的共性和差异通常可以用韦恩图展示。BioVenne能表现出不同组的元素数量以及相同点和不同点,同时展现数据集的大小。[9]
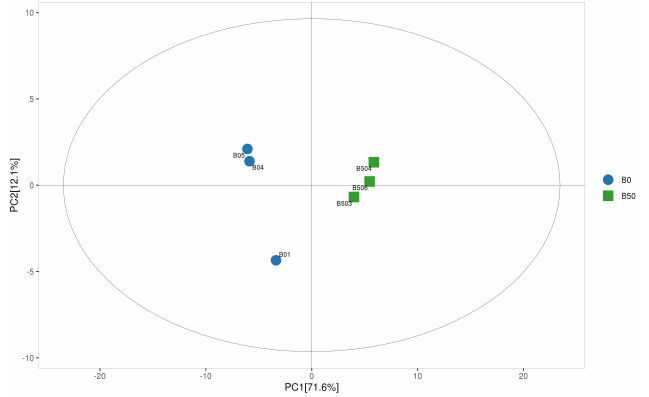
2.1.2 主成分分析[5]
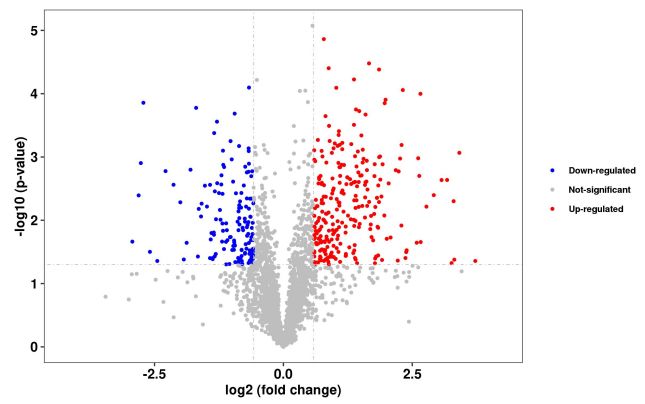
2.1.3 火山图分析[5]
在CHO-K1细胞适应无谷氨酰胺培养基的生长的实验中,用火山图来表示组间差异表达蛋白的表达量变化,阈值设置为fold change < 1.5和p value < 0.05[5](如Volcano Plot-1)。并在图中标注TOP10差异表达蛋白。在实验中,表达量高且显著性高的差异蛋白是容易被关注的,并被选为目标蛋白。这也能降低后续的实验难度。[9]
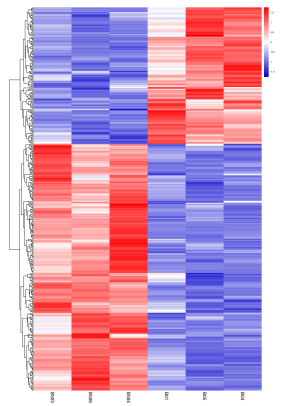
2.1.4 层次聚类分析[5]
2.2 功能分析
2.2.1 GO分析
GO是Gene Ontology的简称,是基因功能国际标准分类体系。它旨在建立一个适用于各种物种的,对基因和蛋白质功能进行注释的信息资源库,他们把基因的功能分成了三个部分,分别是:细胞组分(cellular component, CC)、分子功能(molecular function, MF)、生物过程(biological process, BP)。[4]
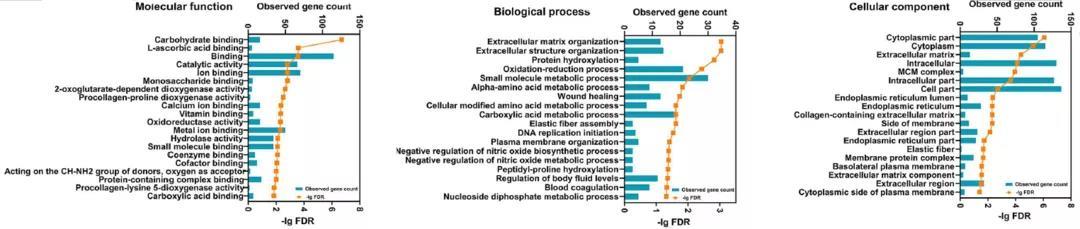
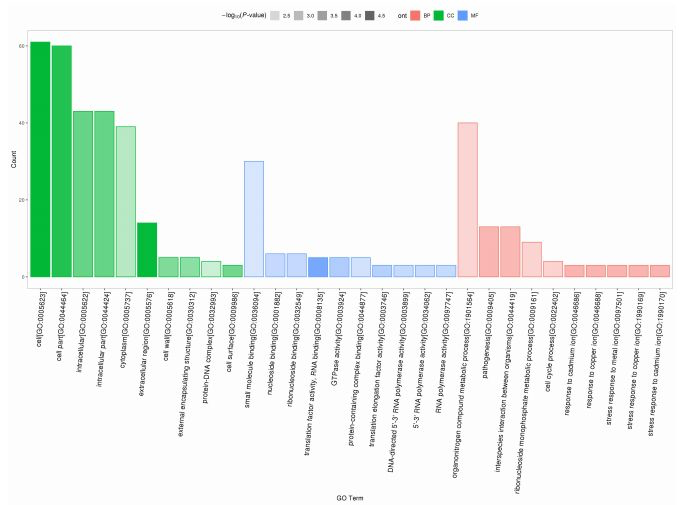
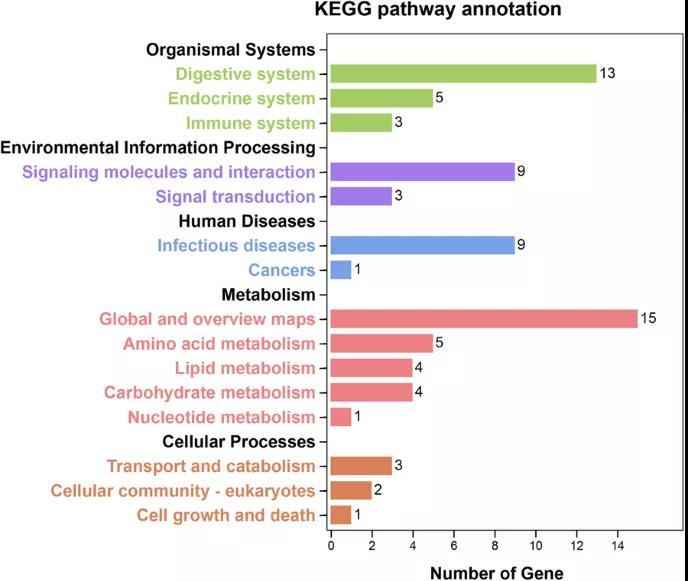
2.2.2 KEGG分析
京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes, KEGG) Pathway数据库 ,储存了基因和基因组的功能信息,包括图解的细胞生化过程如代谢、膜转运、信号传递、细胞周期,以及同系保守的子通路等信息。通过对差异表达蛋白显著富集的信号通路进行分析,可以了解哪些通路在不同实验条件下发生了显著的系统性的改变。[4]
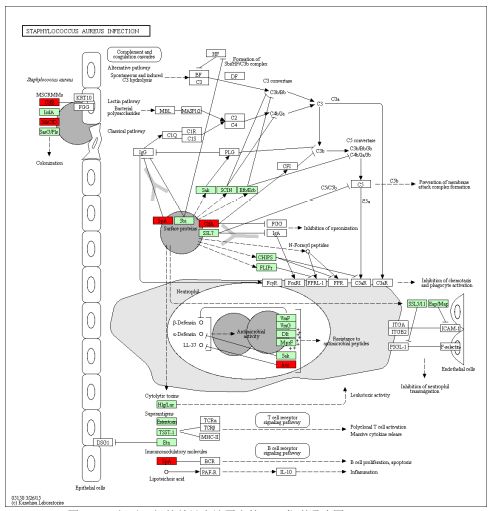
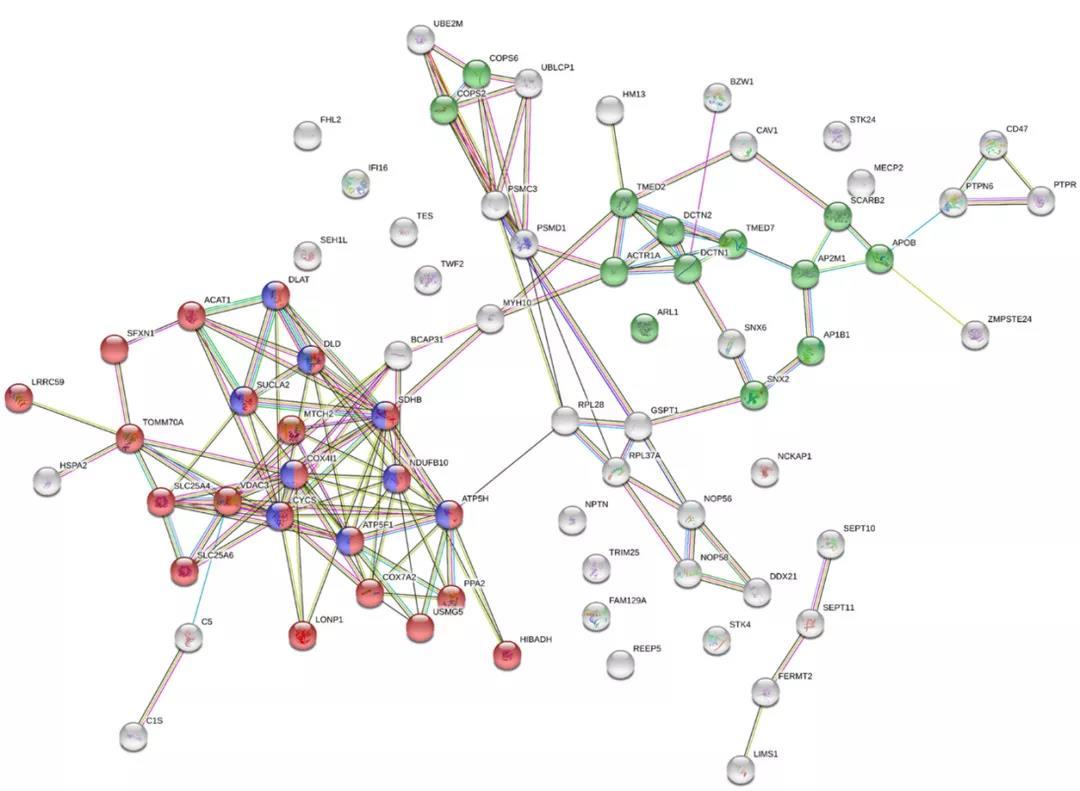
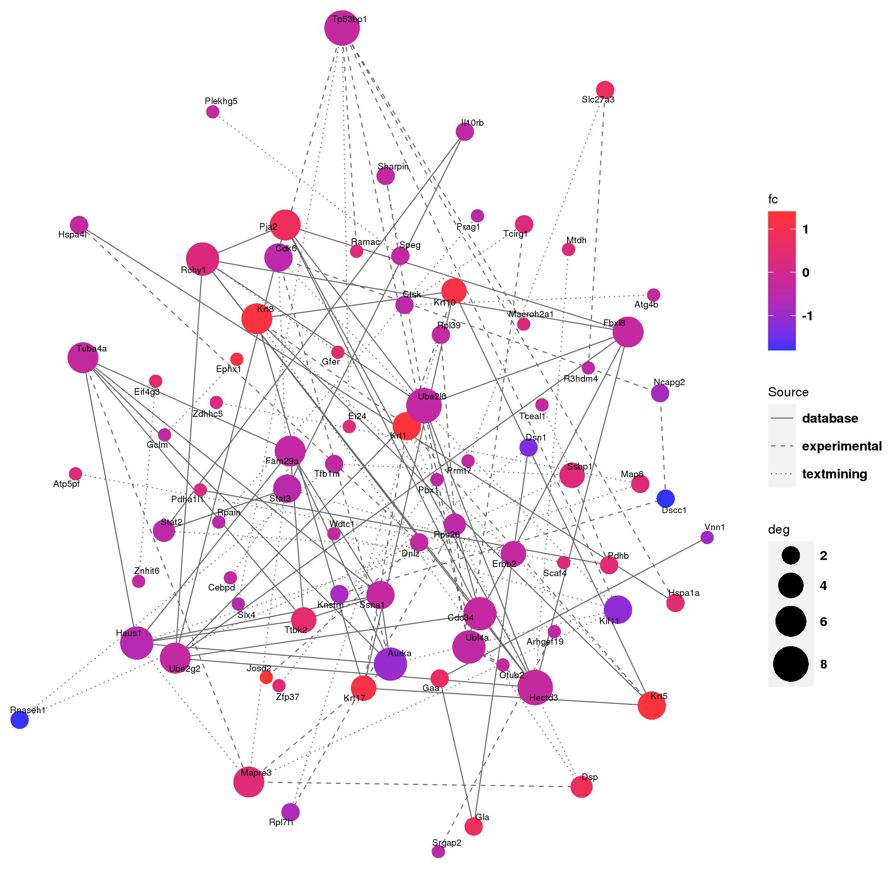
2.2.3 PPI分析
蛋白质-蛋白质相互作用网络分析(Protein-Protein Interaction Network Analysis, PPI Network Analysis),是蛋白质组学的重要研究内容之一。蛋白质在行使生物功能时通过形成PPI网络以维持时间和空间上的协调一致,构建差异表达蛋白的相互作用网络,可以从蛋白质组层面发现差异表达蛋白的变化趋势,进一步帮助我们寻找差异表达蛋白中的关键节点。[4]
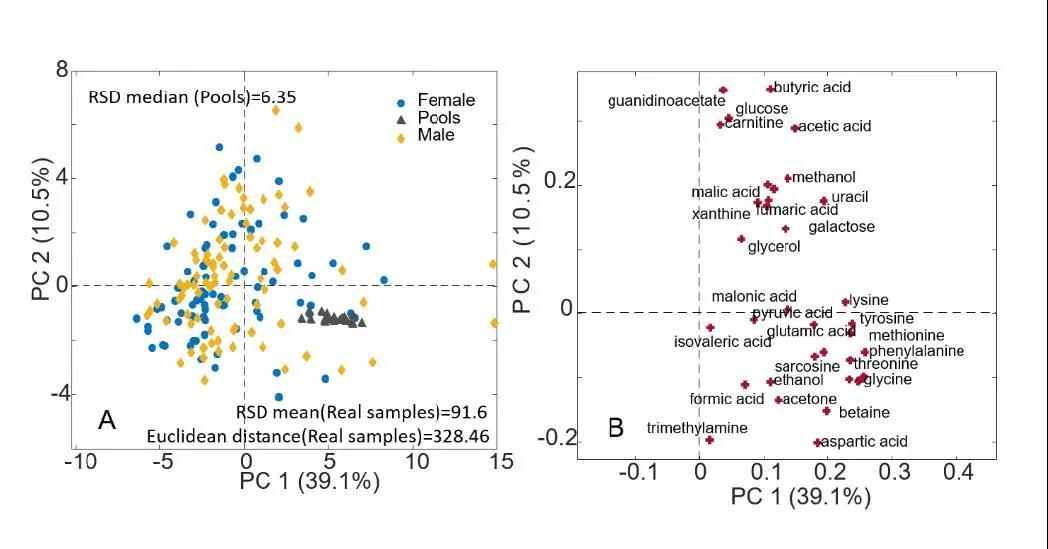
2.2.4 PCA分析[9]
主成分分析 (PCA, principal component analysis)是一种数学降维方法, 利用正交变换 (orthogonal transformation)把一系列可能线性相关的变量转换为一组线性不相关的新变量(也称为主成分),从而利用新变量在更小的维度下展示研究对象的特征。用少数几个主成分的变化来近似代替原来多个变量的变化。通过主成分分析,可以反映不同样本间的关系;对于大量样本,通过离群点,判断是否存在离散样本,在进一步的分析中进行剔除。[9]
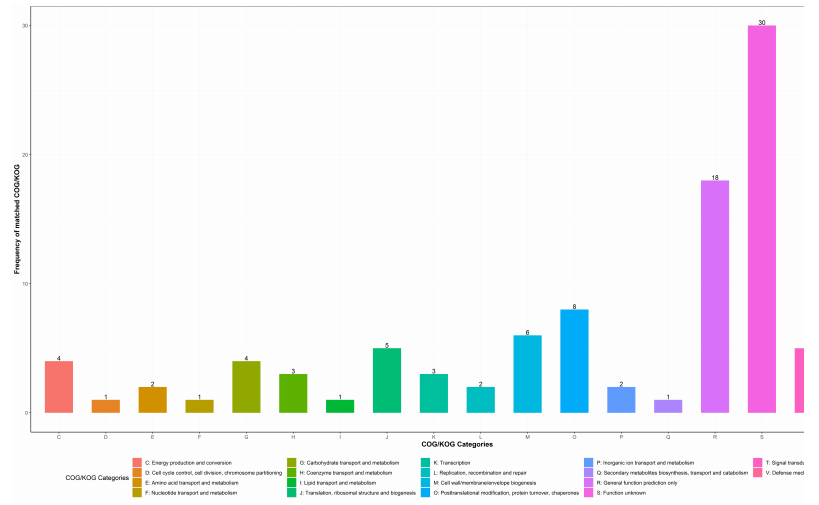
2.2.5 COG 注释分析[5]
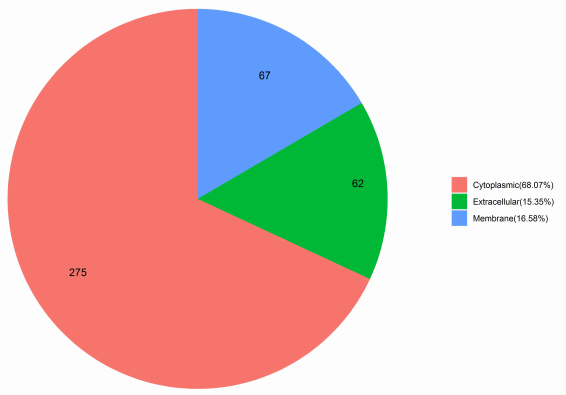
2.2.6 亚细胞定位分析[5]
蛋白质必须转运到其应在的亚细胞结构上才能行使其功能,否则就会出现机体功能紊乱,产生各种疾病。蛋白质的位置是蛋白质最重要的属性之一,有助于确定蛋白质功能,揭示分子交互机理、理解复杂生理过程和开发药物靶标等方面的研究。[9]
2.3 修饰蛋白特有分析
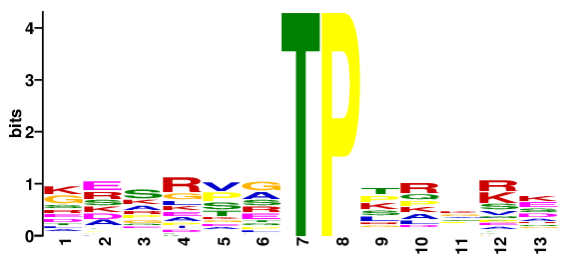
2.3.1 Motif 分析[5]
酶对特定底物的部分生化偏好可能是由修饰位点周围的残基决定的,这种蛋白或多肽序列形成的特定残基模式称为基序(Motif)在蛋白的同源序列中,不同位点的保守程度是不一样的,一般来说,对蛋白质功能和结构影响比较大的位点会比较保守,其它位点则不是很保守。这些保守的位点就称为“模体(motif)”。可以根据蛋白质序列特征(比如基于蛋白质基序)进行功能预测。[9]
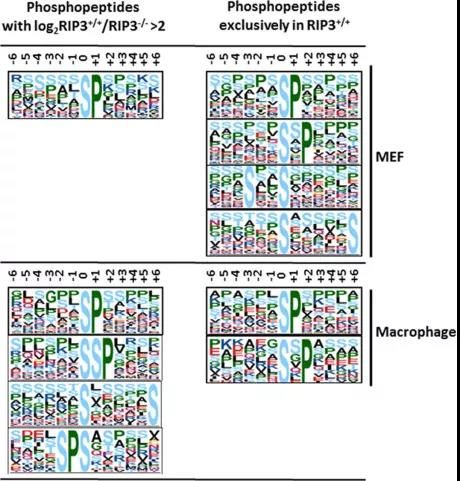
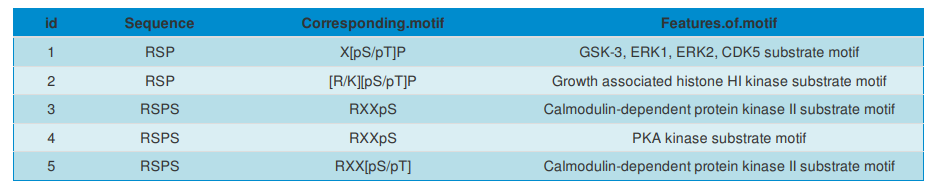
2.3.2 激酶(kinase)预测分析[5]
蛋白激酶(protein kinases,简称PK)。催化蛋白质磷酸化过程的酶。蛋白质的磷酸化过程是神经信息在细胞内传递的最后环节,导致离子通道蛋白及通道门的状态变化。因此,激酶的变化与癌症和疾病密切相关。在磷酸化蛋白组分析中,通常利用motif来预测激酶,揭示参与分子过程的参与者。常用预测激酶网站:http://hprd.org/PhosphoMotif_finder[9]
三、蛋白质组技术流程
3.1 仪器平台[5]
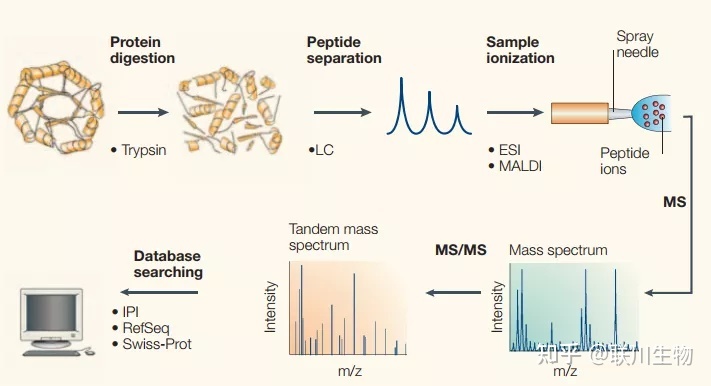
蛋白质的鉴定可以通过其特有肽段的序列来识别,质谱(MS)其扫描速度、灵敏度和分析复杂混合物的能力是一种非常适合分析蛋白质的技术。[11]
其基本原理是通过蛋白质被胰蛋白酶解,然后用液相色谱(LC)的方法对酶解的肽段进行分离,基质辅助激光电离(MALDI)和电喷雾电离(ESI)是常用的对分离肽段离子的检测的方式,然后肽段离子在液相色谱的电离和洗脱,在质谱仪中确定其分子量,质谱仪会进一步分析其碎片的组成。[11]
整个液相和质谱的工作流程就是我们常说的串联质谱(MS/MS),串联质谱识别它特定的氨基酸序列,再通过蛋白数据库及软件的进行分析后,就可以得到蛋白的定性和定量的结果。[11]

3.2 搜索软件[5]
Proteome Discovery、Mascot、SEQUST、MaxQuant、pFind等。
3.3 基本流程[10][11]
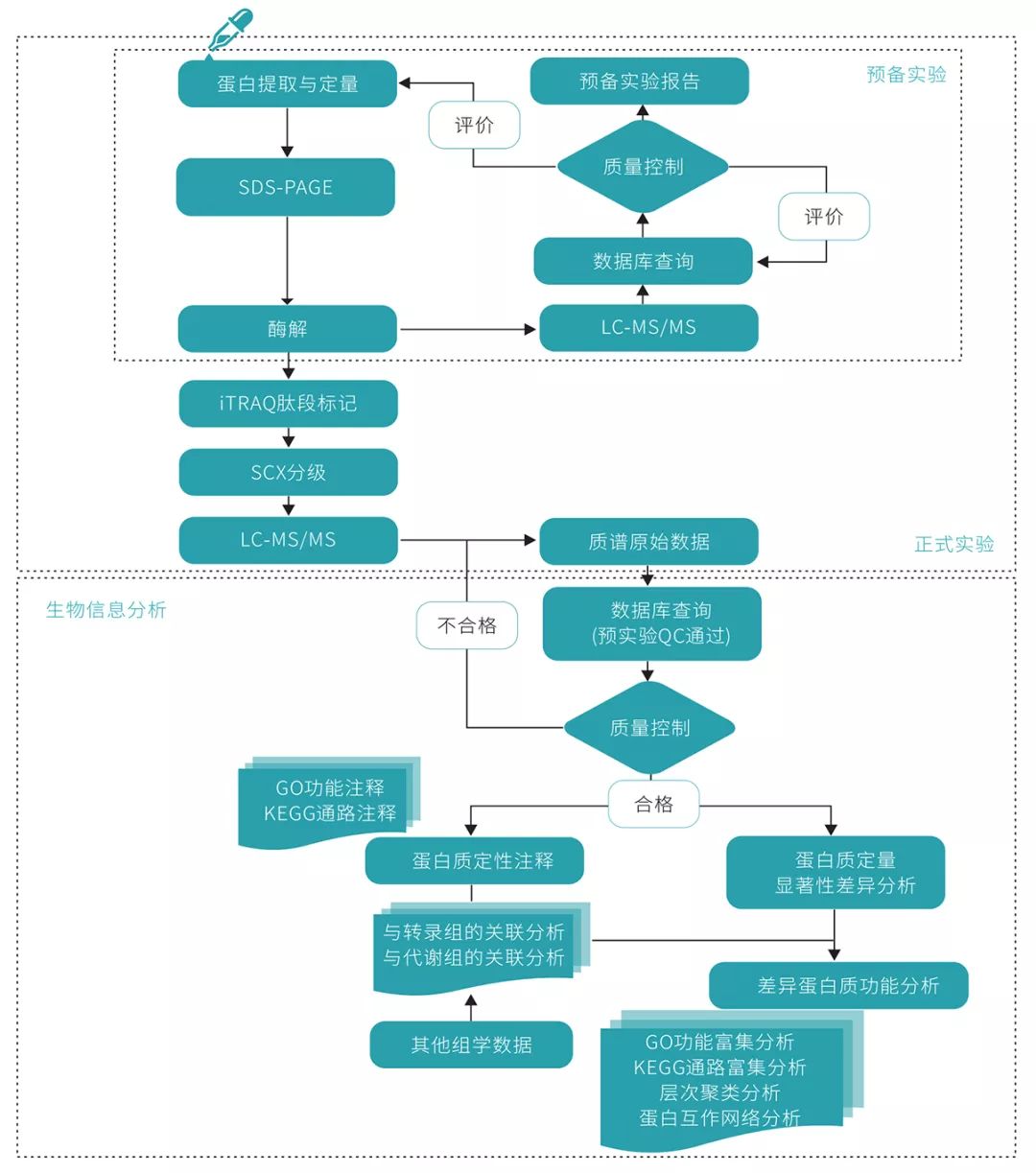
3.4 常见蛋白质组学技术[8][12]
3.4.1 非靶向蛋白质组学
3.4.1 标记定量蛋白质组
(一)ICAT 同位素亲和标签技术[11]
同位素标签技术(isotope-coded affinity tag,ICAT)是一种采用ICAT试剂并用于蛋白质分离的技术,通过具有不同质量的同位素亲和标签(ICATs)标记处于不同细胞中的半胱氨酸,利用串联质谱对复合样本进行质谱分析。
ICAT的方法关键是ICAT实际的应用,该试剂由三部分组成,中间部分称为链接部分,分别是8个氢原子或者重氢原子,前者称为轻链试剂,后者称为重链试剂。
中间部分的一头链接一个巯基的特异反应基团,可以与蛋白质中的半胱氨酸的巯基相连,从而实现对蛋白的标记。另一头连接生物素。用于标记蛋白质或多肽链的亲和纯化。
当采用ICAT技术对蛋白质进行分离和定量时,首先要对蛋白进行标记,然后进行蛋白酶切并对蛋白酶切后的多肽复合物亲和色谱分离,复合物中仅仅被同位素标记的肽段能够进入色谱柱保留,并进行质谱鉴定,其他的大量肽段不被色谱柱保留,通过比较重链和轻链试剂肽段在质谱中的信号强度,可以实现对差异表达蛋白的定量分析。
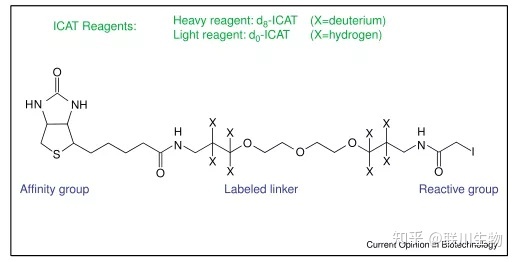
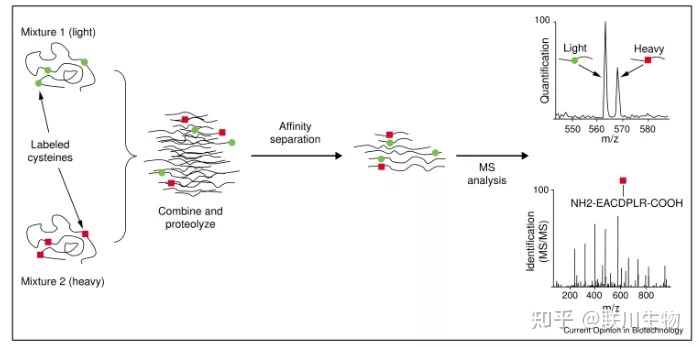
1) 复合物1(轻链)和复合物2(重链)分别用半胱氨酸标记
2) 混合后进行酶切 亲和纯化 后进行质谱分析
ICAT的优点在在于它可以对混合样本(来自正常和病变的细胞或组织)直接检验并能快速定性和定量鉴定低丰度蛋白,尤其是膜蛋白等疏水性蛋白质,可以快速找出重要的参与疾病的标志物等。
ICAT不但可以用来分析整个细胞的蛋白质组,还可以对线粒体一些专门的亚细胞组分进行定量和检测
(二)酶催化 18O 同位素标记法
通常情况下在18O的存在下,酶解蛋白能够产生同位素标记的肽段,最常用的办法是在胰蛋白酶解过程中加入18O,也可以采用Glu-C和Lys-C等中间体的内肽酶,在肽段断裂反应中引入一个氧原子,实现同位素标记。
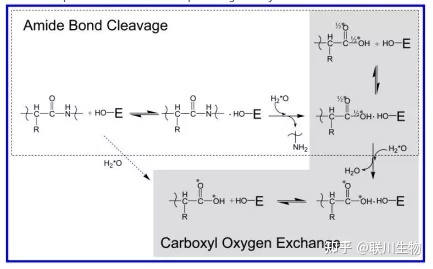
蛋白质水解过程中两种稳定同位素的合并示意图
蛋白质在18O标记的水中被胰蛋白酶解,从而使得18O结合到每个肽段片段的碳端
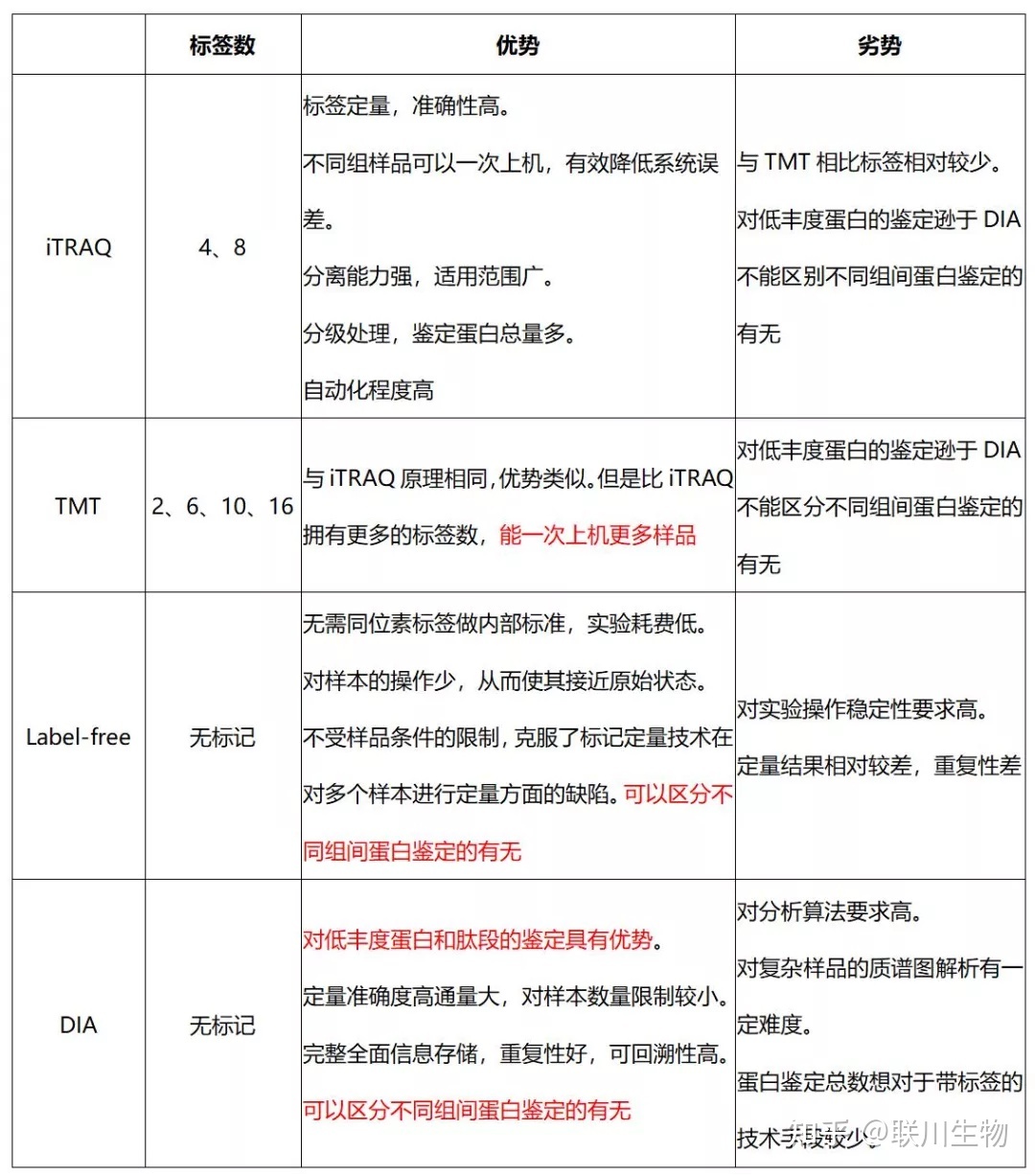
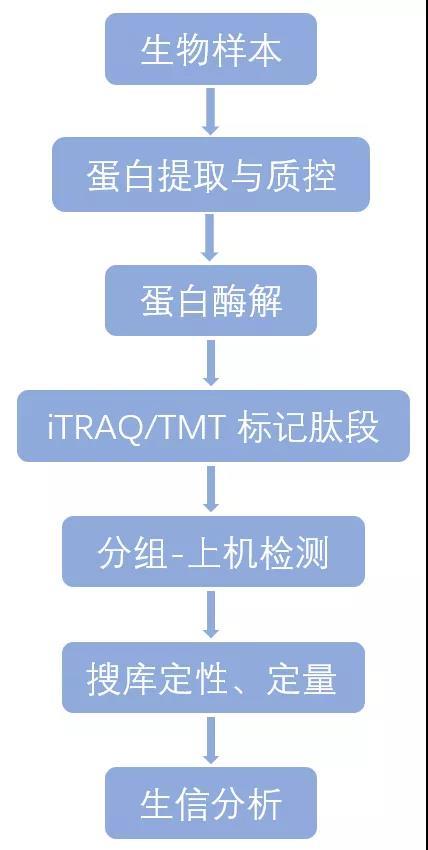
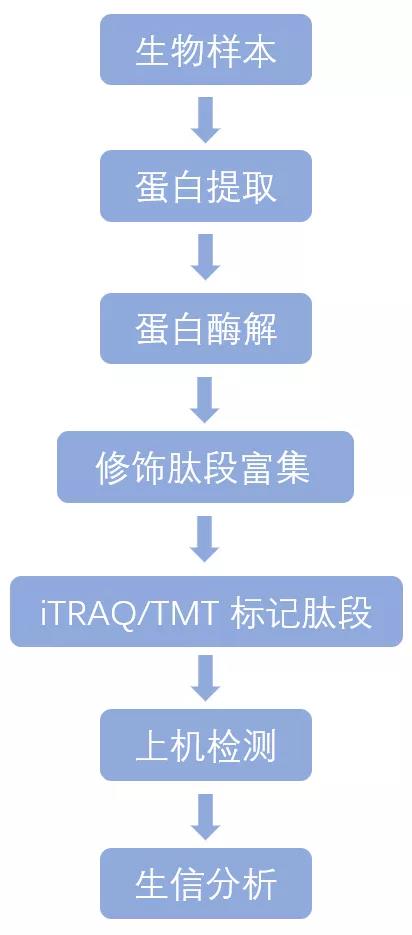
(三) iTRAQ/TMT 标记定量蛋白质组学[8]
iTRAQ 和 TMT 技术采用多种同位素标记,可与氨基反应实现连接,实现多个样本蛋白组的定性与定量。
技术流程:
寄样要求:
- 动物及临床组织标本 100 mg/sample
- 血清、血浆 200 µL/sample
- 细胞、微生物 1x107 cells/sample
- 植物嫩叶、嫩芽 500 mg/sample
- 植物种子、果实 100 mg/sample
- 液氮或者 -80℃ 保存
- 足量干冰运输,避免反复冻融
试剂盒种类:
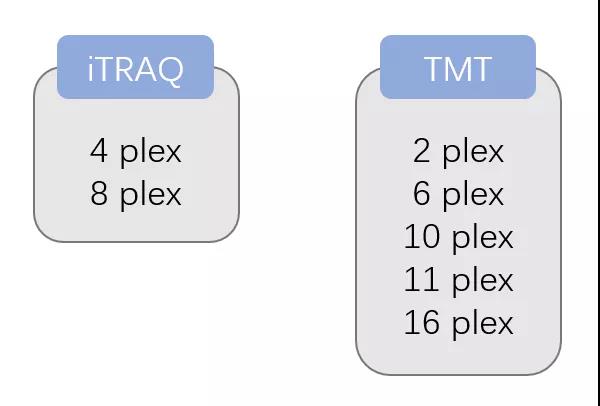
标签区别:
-
ITRAQ

-
TMT
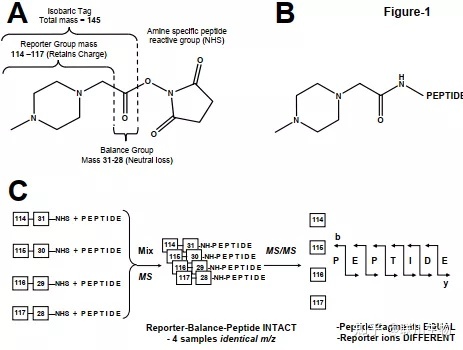
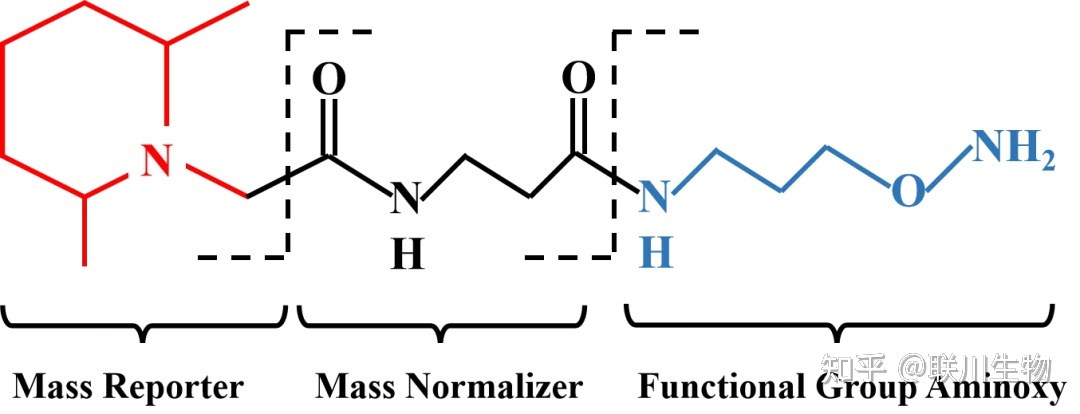
技术优点:
- 适用范围广、高通量、结果可靠
- 灵敏度高
- 分离能力强
- 自动化程度高
3.4.1.2 Label Free 非标记定量蛋白质组学
-
基于质谱无标记定量的蛋白质组学按照定量原理基本上可以分为两大类:一类是基于离子流色谱峰(extracted ion current, XIC)的定量算法,第二类是基于谱图计数法(spectral counting, SC)进行蛋白峰度的相对定量[12]
-
基于离子流色谱峰(extracted ion current, XIC):主要是在保留时间(retention time, RT)轴上,根据肽段母离子的质核比提取不同保留时间下相对应的信号强度并重新计算XIC,利用XIC的面积或信号强度加和作为肽段的定量结果。
- 为了要更准确计算对应的XIC肽段序列有两种办法 1)和精确时间质量标签(accurate masses and time tag, AMT)数据库机型匹配 2)利用蛋白质序列数据库搜库的结果反向确定相对应的肽段序列。
-
基于谱图计数法(spectral counting, SC):根据质谱的原理,一个蛋白的丰度越高,酶切肽段丰度就越高,被质谱检测的概率就越高。
- 将某个蛋白鉴定的蛋白数图谱数作为衡量蛋白峰度的依据,提出谱图记数法,原理简单直观计算也比较方便,也广泛应用到无标定量的方法中。
-
Label Free 技术不依赖同位素标记,可通过液质联用对蛋白质酶解肽段进行分析,分析大规模鉴定蛋白质时所产生的质谱数据,对被检测到的离子峰强度进行积分,以积分面积进行相对定量。
技术流程:
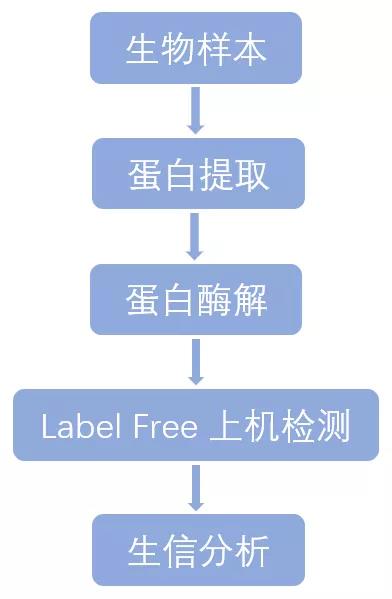
技术优点:
- 无需标记,最大程度的保留样本的真实性
- 可以区分“有”、“无”蛋白
- 不受标签数量的限制,多个样本可以同时进行蛋白定量分析
- 不同物种和不同的样本类型可以同时开展蛋白定量分析
- 处理步骤简单,成本低
技术缺点:
- 低通量
- 定量准确度有限
- 对质谱的稳定性和重现性有很高的要求
3.4.1.3 DIA 定量蛋白质组学
无标记的定量蛋白质组根据实验的需求不同,可以选择不同的质谱数据采集模式,如数据依赖性采集模式(data dependent analysis,DDA)及数据非依赖性采集(data independent analysis,DIA)DDA的数据采集模式。[12]
DDA的采集模式基于鸟枪法的原理,按照离子强度从高进到低采集母离子进行二级碎裂,其结果是有一定的随机性和强度依赖性。[12]
DIA法在2000年首次提出来,它会将整个扫描范围分为若干个窗口,基本实现对所有MS2的信息进行连续性和无偏采集,最大限度的获取碎片离子信息,提高定量的准确度。[12]
数据非依赖性采集(DIA)是一种显著提升蛋白质组学研究通量和可重复性的技术。
经典的 DIA 流程通常依赖于 DDA 谱图库的构建,需要消耗大量的样品,且周期长,成本高。
越来越多的不依赖于 DDA 建库的分析方法相继诞生,例如 DIA-Umpire、PECAN 和 encyclope DIA 等。encyclope DIA 系列技术采用气态在线分离(GPF)。
GPF-DIA 技术流程:
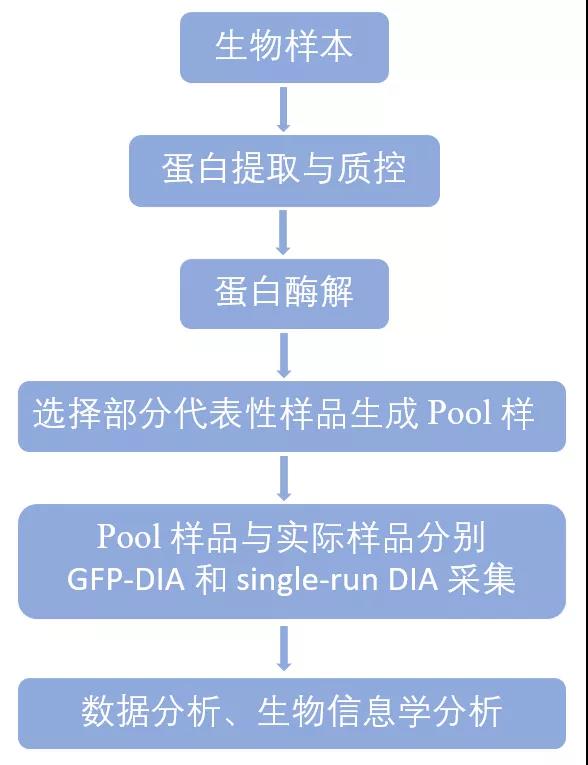
常规 DIA 流程:
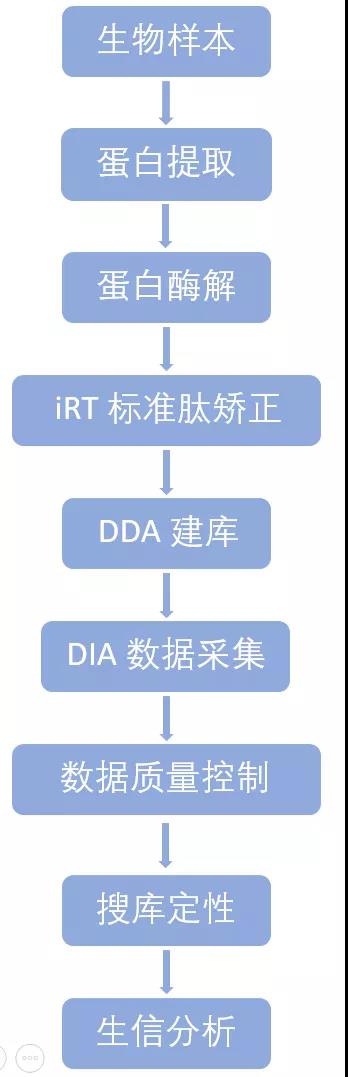
技术优点:
- 采集所有离子及碎片图谱,重现性好
- 基于碎片离子定量,选择性好,可媲美 SPM/MRM
- 循环时间固定,扫描点数均匀,定量准确度高
- 高通量,能同时监测所有目标蛋白
3.4.1.4 定性鉴定蛋白质组学
利用液相色谱质谱联用技术(LC-MS/MS)结合数据库检索的方法,对样本中的混合蛋白进行定性鉴定分析。
技术流程:
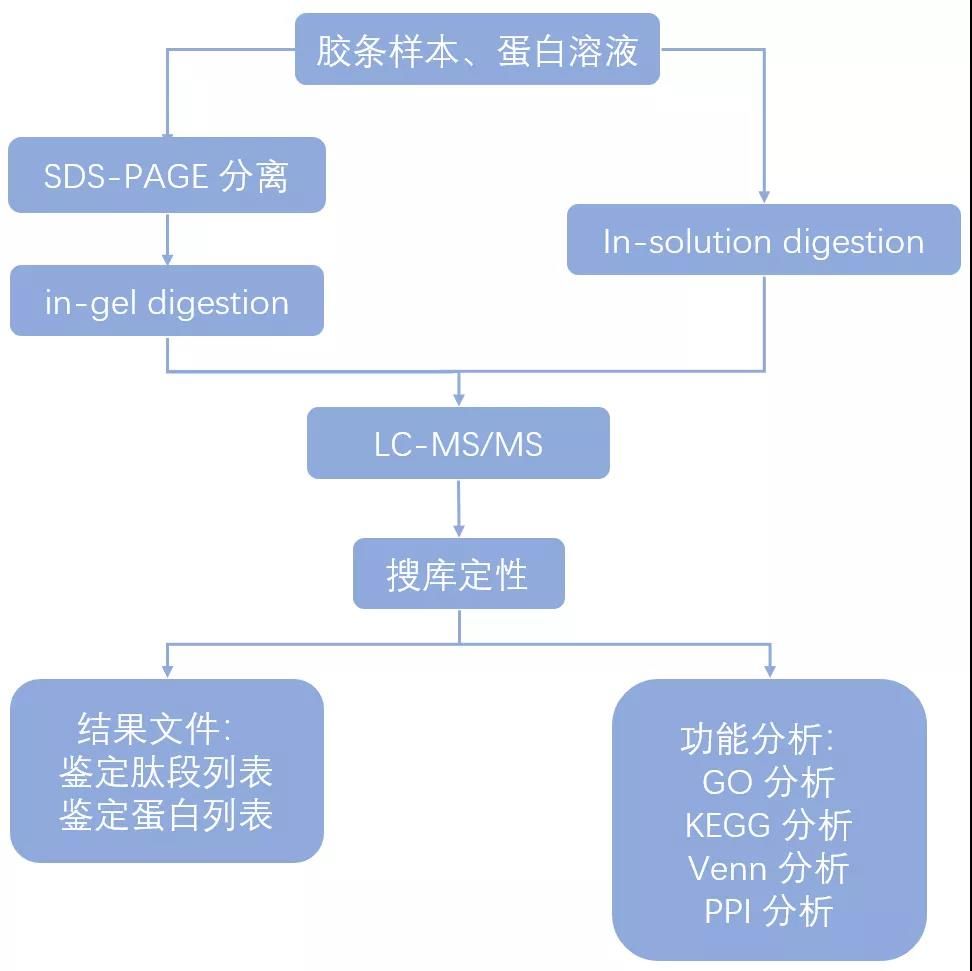
寄样要求:
-
胶条样本
-
考马斯亮蓝染色 SDS-PAGE 凝胶,条带清晰可见
-
条带切取操作过程中佩戴手套,防止角蛋白污染
-
条带置于 EP 管中,-20℃ 保存,冰袋运输
-
-
蛋白溶液
-
需要提供缓冲液体系以及蛋白浓度数据
-
蛋白溶液样本中不要加入 loading buffer
-
液氮或 -80℃ 保存
-
足量干冰运输,避免反复冻融
-
3.4.1.5 多肽组学
多肽组学是以机体内源性多肽和低分子量蛋白质为研究对象,研究多肽组的结构、功能、变化规律及其相关关系。
技术流程:
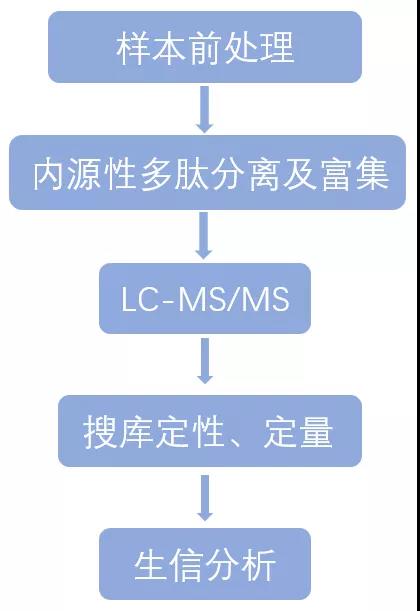
技术优点:
- 高效
- 高通量
- 高特异性
- 破坏度小
- 高灵敏度
3.4.2 靶向蛋白质组学
(PRM 靶向定量蛋白组)
平行反应监测(PRM)是一种靶向检测方法,能够对目标蛋白、目标肽段进行选择性检测,从而实现对目标蛋白/肽段进行相对(绝对)定量。
技术流程:
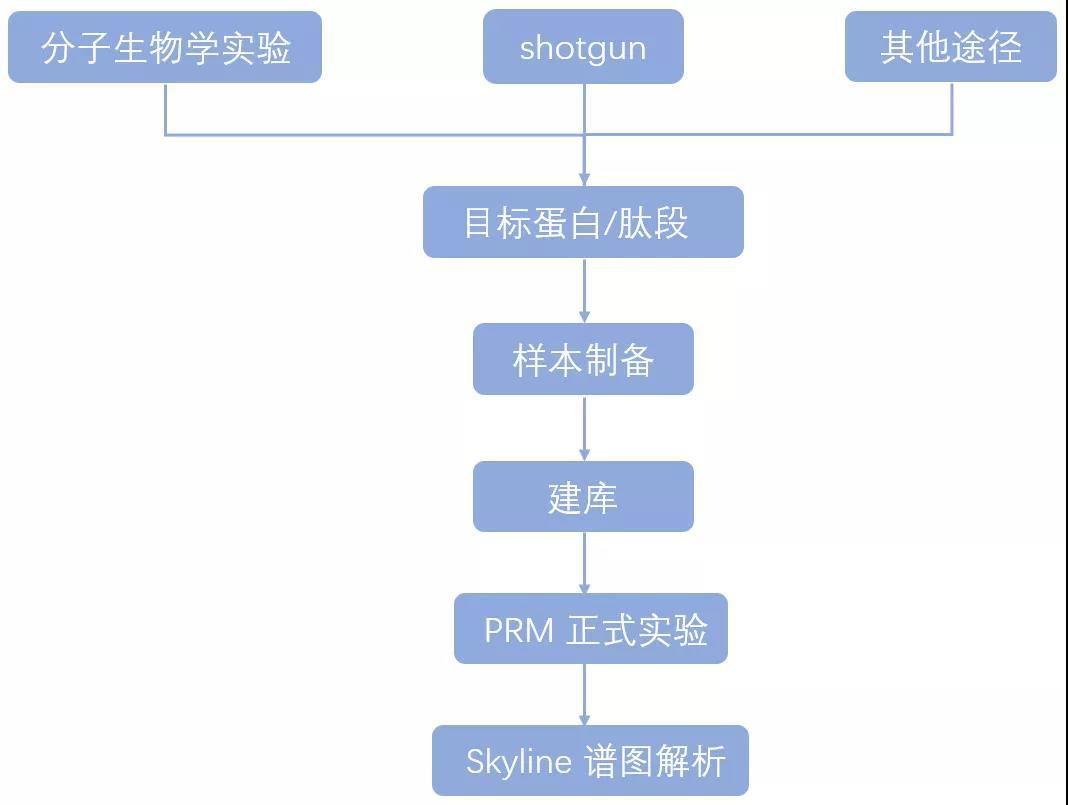
技术优点:
- 不依赖抗体
- 特异性
- 结果准确、可靠
- 高通量
3.4.3 修饰蛋白质组学
利用 LC-MS/MS 结合数据库检索的方法,在搜库时设置修饰搜库参数,实现对目标所有已知的修饰位点磷酸化、乙酰化、糖基化和泛素化进行鉴定。
技术流程:
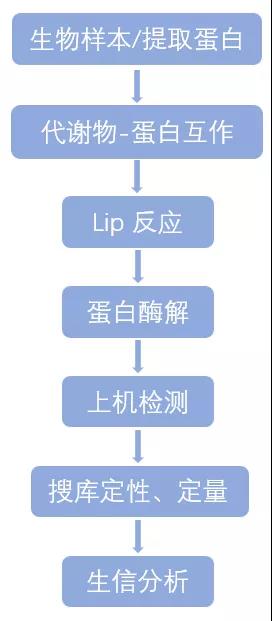
3.4.4 蛋白代谢互作组
代谢物-蛋白质相互作用控制着多种细胞过程,从而在维持细胞稳态中发挥重要作用。MetPro 技术是一种化学蛋白质组学方法,将蛋白质水解(Lip)和质谱(MS)结合起来。用于进行代谢物-蛋白质相互作用的研究,筛选与代谢物具有相互作用的蛋白质。
技术流程:
四、蛋白质组学FAQ[6][7]
Q1:什么样本能做蛋白组学检测?大概可以测到多少的蛋白种类? A:原则上只要能提取到蛋白,就可以做蛋白组检测。样本测到的蛋白种类跟数据库和样本本身蛋白浓度有关,一般数据库越大,可以鉴定到的蛋白质数量越多。也与样本的类型有关,通常,组织细胞相比于体液样本,能够鉴定到更多的蛋白。
Q2:蛋白组学样本寄送前是否需要精确计数或是称量? A:不同样本寄送会有不同的送样要求,按照收集方法提供可以保证实验用量充足。不需要精确计数或是称量,项目开展前会进行蛋白浓度的测定,后续也是不同样本等蛋白量处理、上机检测,但是要保证每个样本的寄样量达到实验要求。
Q3:我的样本不知道有没有降解,量够不够? A:常规蛋白组检测项目,我们会进行蛋白浓度的测定,确定蛋白量是否够用于后续的检测。同时也会进行SDS-PAGE跑胶查看蛋白是否有降解,条带是否明显,如果蛋白量不够或者胶条有异常,会及时跟客户反馈,沟通解决方法。
Q4:哪些样本需要去除高丰度蛋白? A:血清、血浆、脑脊液等体液样本,高丰度蛋白比例达到97%~99%,包括白蛋白、免疫球蛋白、IgG、IgA、抗胰蛋白酶、纤维蛋白原、转铁蛋白等。高丰度蛋白的存在会影响低丰度蛋白的检测,而低丰度蛋白往往是比较关注的生物标记物或者药物靶点,常规是建议去除高丰度蛋白再检测。
Q5:胶条样本如何送样检测? A:可以切关注的部分条带检测,也可以单个样本整个泳道进行检测。如果是整个泳道检测,建议至少分成两份检测,重轻链一份,其他一份,可以增加检测深度。胶条的寄送采用冰袋运输。
Q6:血清血浆样本如何选择? A:都可以选择使用,血浆含有纤维蛋白原,在血清中检测到的蛋白的丰度相对于血浆来说更高一些,一些比较微量的蛋白可能血清可以检测出。但是血清的操作步骤相对比血浆而言更复杂一些,可能会带来人为误差。
Q7:Label Free/TMT&iTRAQ/DIA产品如何选择? A:Label free和TMT&ITRAQ属于数据依赖型采集模式(DDA),通常选择TOP N个母离子肽段进行二级碎裂检测,DIA产品属于数据非依赖型的质谱采集方式,全部母离子都可以进入二级破碎,能够定性到的蛋白数量可能会更多,依赖于建库数据;
与标记蛋白组相比,DIA和label free均不会受限于样本数量;与label free技术相比,DIA的可重复性更高。建议样本数量比较大的时候选用此技术。样本数量较小的时候,如果样本之间差异比较大,需要查看蛋白的有无信息,推荐做label free产品。
如果样本例数不多,想进行深度检测,定性定量更准确,建议标记定量产品。
Q8:标记产品最多可以做多少样本? A:目前标记试剂盒主要分为TMT/iTRAQ两种,试剂盒一次最多可以标记16个样本,如果超过16例,可以通过搭桥样本来实现批次间的校正。
Q9:物种如何选择数据库? A:常规是使用uniprot网站该物种的蛋白数据库作为搜库文件,如果该物种的数据库比较小或者没有数据库,可以选择以下三种解决方案:(1)扩大物种的层级,选择上一层级或是更大层级的物种库;(2)选择近源物种库;(3)转录组测序结果作为数据库。有些个别的物种有专门的数据库网站也可以采用去进行搜库分析。
Q10:常规定量蛋白组实验的主要步骤有哪些,主要采用哪些仪器检测?数据分析内容包含哪些? A:实验步骤:首先获得生物学样本,然后进行蛋白提取,定量,SDS-PAGE跑胶质控,还原烷基化,酶解,获得肽段,(标记+分组分),上机检测,搜库软件搜库,数据分析;
仪器:主要采用Q Exactive™ HF-X 、Orbitrap Fusion™、Orbitrap Exploris™ 480等;
数据分析:主要分为基础数据分析、⾼级数据分析和个性化数据分析三个部分。基础分析主要包括差异表达蛋白筛选、层次聚类分析分析、COG注释分析、亚细胞定位分析、GO注释富集分析、KEGG注释分析、PPI分析等。
Q11: 蛋白组一般是建议做多少个生物学重复? A: 原则上是生物学重复越多越好,排除个体差异,筛选的差异蛋白更准确,验证成功率更高。考虑到经费、统计分析需要及后续编辑可能会质疑的情况,临床样本建议每组十个重复以上,其他来源样本每组至少三个重复。
Q12: 蛋白组样本如何寄送? A: 常规组织、细胞、体液等生物样本需要低温保存,干冰寄送。胶条样本可以冰袋寄送。
Q13:蛋白组学能否测未知的蛋白?或者是表达的外源蛋白,该样本物种数据库里没有的蛋白能否测到? A:蛋白组学检测结果是与已知的数据库蛋白进行比对,无法预测未知的蛋白,如果需要检测未知蛋白,可采用测序等其他方法。如果数据库里不包含关注的蛋白,那么无法比对到。可以将关注的蛋白序列加入到数据库里作为搜库文件进行分析。
Q14:磷酸化蛋白组学实验的流程? A:磷酸化蛋白组可以做非标记产品,也可以做标记产品。目前标记产品做的较多,可以增加检测深度。实验流程主要是包括样本前处理、蛋白的提取、蛋白浓度的测定和跑胶质控、酶解成肽段、修饰肽段的富集、(肽段标记)、质谱检测。方案多样化,可以进行选择,可以先富集再分级再检测,或者先标记、分级、每级富集等等。
Q15:蛋白检测结果较少是为什么呢? A:首先可能是数据库比较小的原因,导致检测到的结果比较少。可以选择扩大层级或者选择研究较多的近缘物种、模式物种数据库进行搜库分析。其次看下胶图,判断样本本身条带是否较少,是否存在高丰度蛋白,高丰度蛋白的存在会影响检测数量。
Q16: 鉴定到的蛋白与凝胶电泳图谱中估测分子量差异很大的原因?或者为什么SDS-PAGE胶上不同位置条带的质谱鉴定结果中含有同一个蛋白? A: 由于体内或者体外因素,导致同一蛋白存在不同形式的修饰、剪切或者降解,从而形成凝胶电泳图中能够看到的存在差异分子量的蛋白条带。然而这些蛋白在质谱鉴定时会同时指向数据库中同一个、全长的、无修饰的蛋白理论序列。因此会出现凝胶电泳图中的蛋白分子量(实际分子量)与鉴定的蛋白分子量(理论分子量)不同的情况。
Q17: 差异蛋白如何筛选? A: 差异蛋白筛选的条件主要是结合T检验的p值和FC值,一般标记产品按照FC>1.2 or FC<0.83,p<0.05;非标记产品按照FC>1.5 or FC<0.67,p<0.05进行筛选。实际过程中也会结合检测结果进行放宽或者卡严,一般控制在检测结果20%以内,5-10%为佳。
Q18: 做完蛋白组学,选用何种方法验证呢? A: 常规蛋白验证方法主要包括WB、ELISA、PRM。如果关注的蛋白数量不多且有对应的商品化检测试剂盒或者抗体,建议选择ELISA或WB方法进行验证,此方法更为成熟。如果关注的蛋白数量较多,也没有商品化的抗体,建议选用PRM的方法。经费充裕的情况下,抗体制备也是一个不错的选择。
Q19: 通过western blot检测到的蛋白,为什么质谱没有检测到或者只检测到一个肽段? A: Western blot 检测是将目的蛋白信号放大很多级之后进行检测的,灵敏度很高,(除非特异性结合之外)几乎不受复杂样品中背景蛋白丰度的影响。质谱检测时样品中丰度较高的蛋白优先、多次被检测到,而丰度较低的蛋白则因为肽段信号过弱被淹没而不能被检测到。因此,如果样品中待检测的目标蛋白丰度较低,即使WB能够检测到,质谱并不一定能检测到或者仅能检测到较少的肽段数。
Q20: 同一批样本,转录组数据下调,但是蛋白组学结果上调,这种是什么情况? A: 这是正常的现象,上下游不是一一对应的关系,常规mRNA与蛋白质间的相关系数仅为0.4~0.5。一个蛋白的表达受到很多因素的控制,除了这个蛋白对应的mRNA之外,还有比如转录因子,增强子,抑制子,DNA和RNA的修饰作用等等。
参考资料
[1]:生物信息学 | 数据库
[2]:查询蛋白质信息 | 网站?
[4]:生信分析 | 蛋白质组学数据分析图形展示系列之常见分析图
[6]:上分攻略1 | 蛋白质组学FAQ
[10]:蛋白质组学 | 技术介绍|
[11]:干货分享 | 三个蛋白组学数据库的应用





暂无评论内容